Infectious Diseases
Research Areas | Product Areas | Full List of Infectious Disease Targets | Background | Related Information
Infectious diseases are caused by pathogenic microorganisms such as bacteria, viruses, fungi or protozoa. Infectious diseases can spread via direct or indirect contact. Infections can be transmitted from animal to person (zoonotic) and from person to person. Diseases can spread through exchange of bodily fluids from sexual contact and during blood transfusion. It is also possible to contract an infectious disease from an inanimate object which has previously been contaminated with pathogen. Some infectious organisms live naturally in the surrounding environment but cannot be transmitted from person to person. Examples of these pathogens include anthrax (bacterial) and histoplasmosis or blastomycosis (fungal). R&D Systems offers tools for the study of Human Immunodeficiency Virus (HIV), Herpes virus, Influenza virus, and many more infectious diseases.
Featured Research Areas and Resources
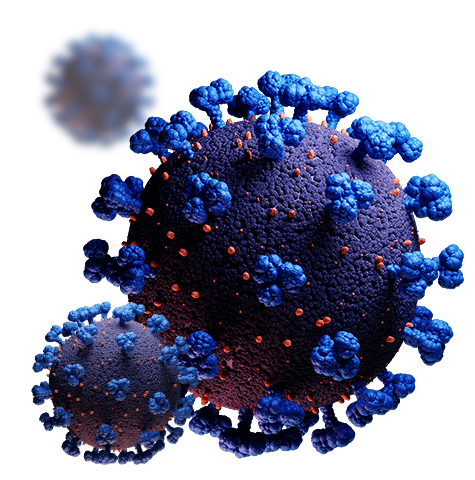
Coronavirus
Browse antibodies, proteins, and small molecules to advance your research into SARS-CoV-2.
Zika Virus and Other Flaviviruses

Learn more about R&D Systems immunoassays and applications for viral entry and replication of Zika virus and other Flaviviruses.
RIG-I-like Receptor (RLR) Pathway
Explore the role of RIG-I-like Receptor (RLR) in virus detection and initiation of the innate immune response.
Infectious Disease Research Product Areas
| Antibiotics | Antimalarials | Fluorescent Probes for Imaging Bacteria |
| Antifungals | Antivirals | Prion Protein |
Mechanisms of Infection
Infections can spread from person to person via direct or indirect transmission. Examples of direct transmission include via exchange of bodily fluids from sexual contact or blood transfusion. Infections can be indirectly transmitted via air borne droplets, as a result of sneezing, coughing or talking, or from an inanimate object that has previously been contaminated with a pathogen. In addition, some infections are transmitted from animal to person (zoonotic), such as ebola, swine flu (H1N1), rabies and COVID-19 (SARS-CoV-2).
Response to Infection
The immune system is a protective mechanism which responds to pathogenic invasion by mounting an immune response. It can discriminate between self and foreign antigens; this enables an organism to maintain homeostasis. If this ability to discriminate is disrupted, the immune system can initiate an autoimmune reaction against cells or tissues of the body, which can develop into an autoimmune disease.
Innate Immune Response
Innate immunity is an organism's first line of defense against invasion by any pathogen, including those not previously encountered and is not specific to a particular invader. The innate immune system consists of physical barriers, such as the skin and mucus membranes, which block entry of foreign particles. If these surfaces are penetrated, immune cells, including monocytes, macrophages, neutrophils, dendritic cells and natural killer cells, are activated which respond by phagocytosis of the invader and release of cytokines and complement to recruit further cells and amplify the inflammatory response. Innate immunity is evolutionarily conserved.
Adaptive Immune Response
Adaptive or acquired immunity is an organism's response to a specific antigen, pathogen or immunogen and is triggered by the body's recognition of an invader that it has encountered previously. The adaptive immune system consists of an antibody response and a cell-mediated response. The cells comprising the adaptive immune system are known as lymphocytes, of which there are 2 types: B cells and T cells. Upon activation B cells release antibodies which bind specific antigens, inactivating viruses or bacterial toxins and targeting them for destruction by the innate immune system. Some T cells, known as CD8+, act as killer cells, directly attack virus-infected cells that present viral antigens on the surface. Other T cell populations known as CD4+ or helper cells (Th), can signal to other immune cells such as macrophages to destroy an invading pathogen. A third population T cells, termed regulatory T cells or Tregs, distinguish alien cells from self, preventing the immune system from mounting an inappropriate immune response. However, Tregs also have a role in cancer as they can prevent the immune system from recognizing tumor cells as abnormal.
Confidently Test with Armored RNA Quant® Molecular Controls from Asuragen, a Bio-Techne brand.
Related Information
- ACE-2: The Receptor for SARS-CoV-2
- Bacillus anthracis vs. Dendritic Cells
- Blog: RSV And Influenza Viruses Take Center Stage As Early Infections Spike
- Cutaneous Immunology & Infectious Disease
- Ephrin-B2: A Receptor for Henipaviruses
- Host Defense/Innate Immunity
- Significant Events in the History of Virology