The ERK Signal Transduction Pathway:1
Its Role in Growth Factor Signaling and Cancer
Contents
Growth factors recruit a large network of signaling proteins through receptor tyrosine kinases, to execute their cellular programs.
The first of these networks to be discovered was the Ras-Raf-ERK signal transduction cascade, defined by Extracellular Signal-regulated Kinase-1 (ERK1) and ERK2.1 One of four Mitogen-Activated Protein Kinase (MAPK) signaling pathways, the ERK phosphorylation cascade's importance in intracellular signaling has been compared to the role of the Krebs cycle in energy metabolism.2 The ERK cascade functions in cellular proliferation, differentiation, and survival, and its inappropriate activation is a common occurrence in human cancers.
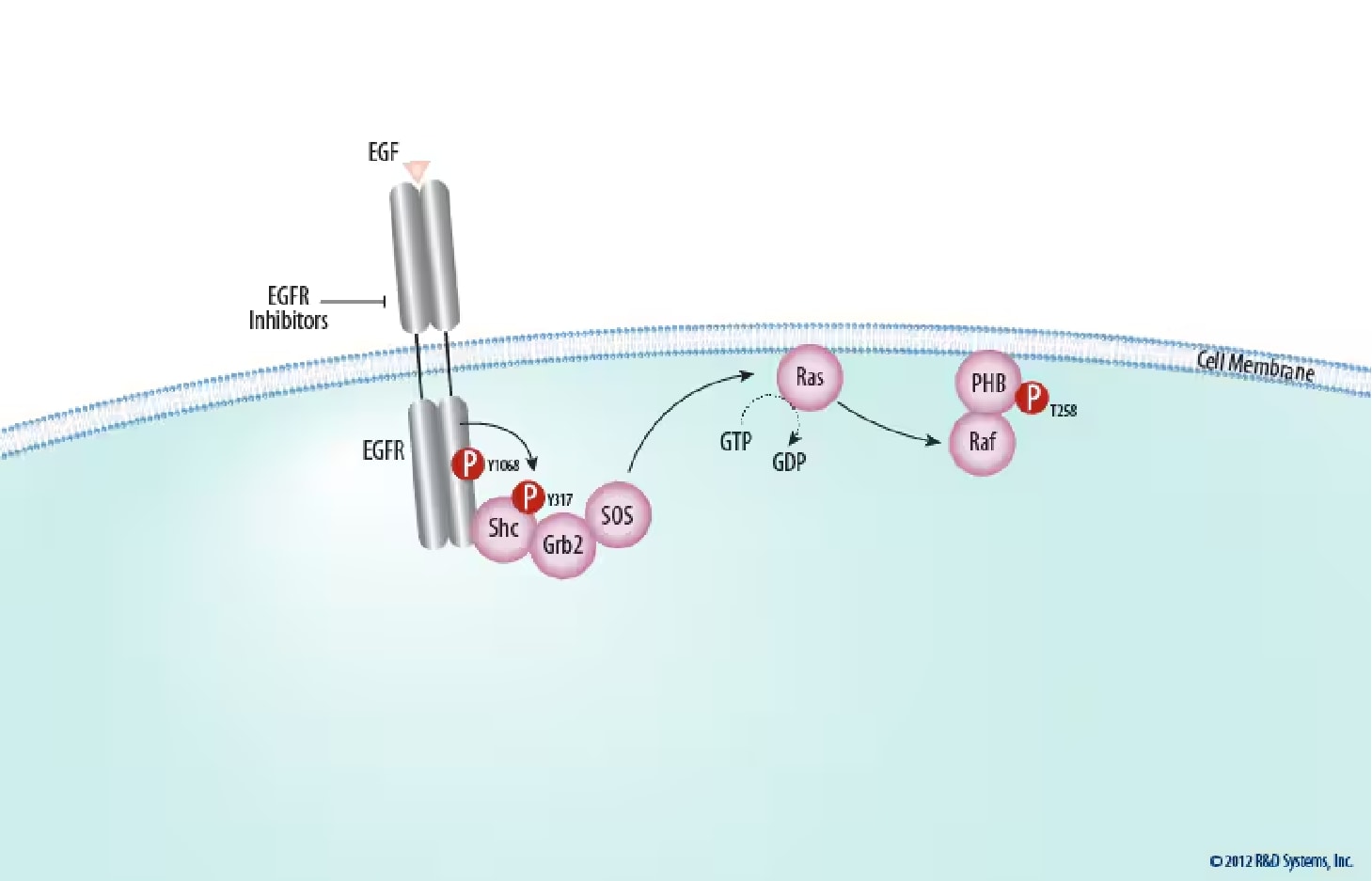
Figure 1. Ras Activation at the Plasma Membrane. The activated membrane-spanning epidermal growth factor receptor (EGF R) becomes a platform for the assembly of a signaling complex that includes the cytoplasmic growth factor receptor bound protein 2 (Grb2) and son of sevenless (SOS), which activates the membrane-bound GTPase, Ras. Ras subsequently activates Raf kinase, which is recruited to the membrane through its interaction with Prohibitin (PHB). EGFR activation can be blocked pharmacologically; Find EGFR inhibitors offered by Tocris Biosciences.
Upstream from ERK
During growth factor stimulation, the ERK phosphorylation cascade is linked to cell surface receptor tyrosine kinases (RTKs) and other upstream signaling proteins with known oncogenic potential (Figure 1).3
RTKs
Membrane spanning cell surface receptors of the RTK family are endowed with intrinsic tyrosine kinase activity, catalyzing the transfer of the gamma-phosphate of ATP to the hydroxyl groups of tyrosine residues on target proteins. All RTKs contain a frequently glycosylated extracellular ligand binding domain, connected through a single transmembrane helix to the cytoplasmic domain. The cytoplasmic domain contains a conserved protein tyrosine kinase (PTK) core and regulatory regions that are subjected to autophosphorylation and phosphorylation by other kinases.4 Nearly all RTKs are monomers at the cell membrane, with ligand binding or ectopic overexpression resulting in receptor dimerization and tyrosine residue autophosphorylation in trans.
As the first RTK to be discovered,5 the Epidermal Growth Factor Receptor (EGF R), also known as v-erb-B Transforming Protein of an Avian Retrovirus 1 (ErbB1), has helped establish many of the principles of RTK function.5, 6 EGF R forms an RTK subfamily with three other closely related receptors: the orphan receptor ErbB2, also known as Human EGF Receptor 2 (HER2) and neu; ErbB3 (also known as HER3 and characterized by an impaired kinase domain); and ErbB4 (also known as HER4).7 Activating mutations and transforming overexpression, mimicking receptor oligomerization of EGF R and its fellow family members, have been implicated in numerous cancers, including mammary carcinomas, squamous carcinomas, and glioblastomas, and will be our model RTK here for further analysis.3
Adaptors
The EGF receptor contains at least 20 tyrosine residues capable of phosphorylation in its cytoplasmic domain, and seven of these are autophosphorylation sites.8,9Tyr autophosphorylation sites on EGF R and other RTKs provide a mechanism for the recognition and assembly of signaling complexes, functioning as binding sites for Src Homology 2 (SH2) and phosphotyrosine binding (PTB) domains of a variety of signaling proteins.4 Therefore, activated RTKs become a platform for the recognition and recruitment of a specific complement of signaling proteins. One such signaling protein is Growth Factor Receptor Bound Protein 2 (GRB2). GRB2, a cytosolic adaptor, contains a central SH2 domain flanked by two Src homology 3 (SH3) domains that allow it to constitutively associate with the proline-rich regions of the nucleotide exchange factor Son of Sevenless (SOS).10 Phosphotyrosine 1068 of the activated EGF R is a binding site for the SH2 domain of GRB2, either directly or through the assistance of another SH2 adaptor, Src Homology 2 Domain Containing Transforming Protein (Shc).11,12 Activated EGF R phosphorylates Shc on Tyr317, which promotes the interaction between Shc and GRB2 while also reducing the binding affinity of Shc for EGF R.13 The recruitment of GRB2 from the cytoplasm to the plasma membrane brings SOS near the membrane-bound product of proto-oncogene c-Rat Sarcoma Viral Oncogene Homolog (ras). Through guanine exchange, SOS enhances GDP release and GTP binding to Ras, converting this GTPase into its active conformation.
Ras
Ras is a notable member of the large family of GTPases, proteins that bind and hydrolyze GTP. First discovered as transforming oncogenes of murine sarcoma viruses, three highly related 21 kDa mammalian proteins, Harvey-Ras (H-Ras), Kirsten-Ras (K-Ras), and Neuroblastoma-Ras (N-Ras) have been identified.14 Activating mutations of these Ras isoforms, which impair GTPase activity and stabilize the GTP bound state, or of their downstream effectors, are found in nearly one-third of all human cancers, making these oncoproteins among the most potent transforming polypeptides known.15,16
Ras family members are anchored to the cytoplasmic face of the plasma membrane by C-terminal farnesylation. This localization to the inner leaflet brings Ras into close proximity with SOS, stimulating the exchange of GDP bound to Ras with GTP from the cytosol. This exchange activates Ras conformationally, allowing it to interact with a number of downstream effectors.15 Within the ERK signaling cascade, active Ras functions as an adaptor that binds to effector Raf Murine Sarcoma Viral Oncogene Homolog (Raf) kinases with high affinity, causing their translocation to the cell membrane, where Raf activation takes place via Prohibitin (PHB).18,19
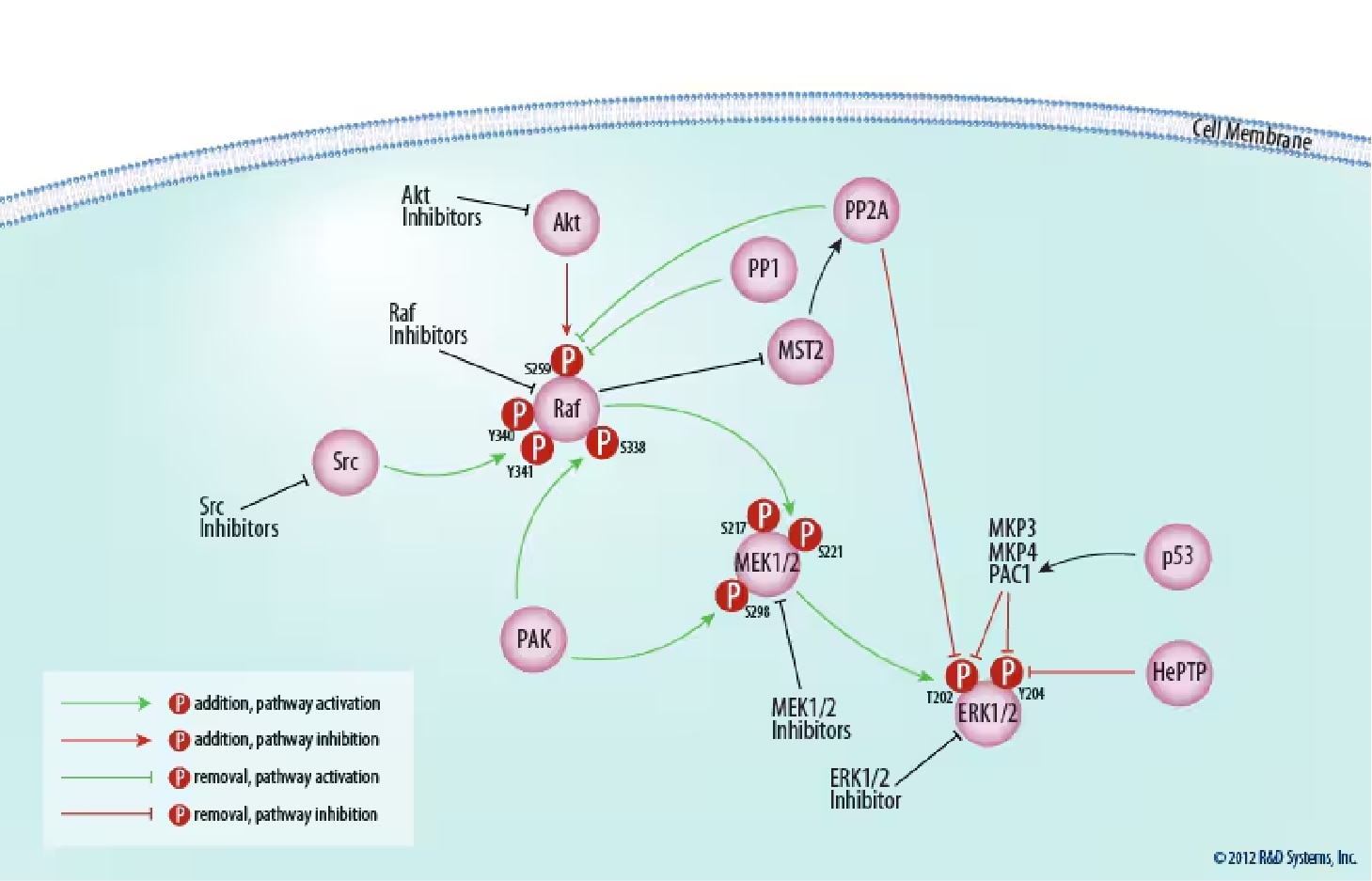
Figure 2. Regulation of the Raf/MEK/ERK Core Signaling Module.The signaling cascade from Raf to ERK is regulated both positively and negatively by multiple kinases and phosphatases; see text for details. The activity of many of these molecules can be modulated pharmacologically. Click on each of the following molecules to view inhibitors/activators offered by Tocris Biosciences: Akt, Raf, Src, MEK1/2, ERK1/2, and PP1 and PP2A.
The Core Module
Activation of all MAPKs is regulated by a central three-tiered core signaling module, comprised of an apical MAPK kinase kinase (MAP3K), a MAPK kinase (MEK or MKK), and a downstream MAPK (Figure 2).
Raf
Like Ras, the MAP3K Raf was first discovered in the form of a mutant retroviral transforming agent, v-raf.20 Raf is a serine/threonine protein kinase, catalyzing the phosphorylation of hydroxyl groups on specific serine and threonine residues.21 Mammals possess three Raf proteins, ranging from 70 to 100 kDa in size: Raf-1, which is ubiquitously expressed; A-Raf, found in cartilage, intestine, heart, spleen, thymus, cerebellum, and urogenital tissues;20 and B-Raf, present in multiple isoforms and strongly expressed in fetal brain and adult cerebrum.22,23,24 In addition to their different cell-specific expression, the phenotypes of raf knock-out mice also vary. A-raf deficient mice survive birth with intestinal and neurological abnormalities, while mice with a targeted disruption of the B-raf gene die of vascular defects during mid-gestation.25,26 Raf-1-/- mice also die in utero with a number of developmental defects.27 These results indicate that Raf-1 may serve a general role in tissue formation, whereas A-Raf and B-Raf fulfill more specialized duties.
Recruitment to the plasma membrane by GTP-bound Ras is the initiating event in Raf activation. The effector domain of Ras binds Raf at two locations in the MAP3K's N-terminus, the Ras-binding domain (RBD) and the cysteine-rich domain (CRD), with binding at both sites necessary for activation.28 Additionally, it has been shown that Prohibitin phosphorylated on Thr258 and Tyr259 associates with Raf-1, v-Akt Murine Thymoma Viral Oncogene Homolog (Akt), and Ras at the plasma membrane and is essential for Raf-1 activation.29 Different Ras isoforms appear to activate Raf with varying ability, despite binding in vitro with comparable affinity. For example, K-Ras both recruits Raf-1 to the plasma membrane more efficiently, and activates the recruited Raf-1 more potently than H-Ras.30 It has also been suggested that B-Raf is the primary target of oncogenic Ras isoforms.31 Activating mutations of B-raf have been reported in approximately 50% of malignant melanomas, all within the kinase domain and most being a missense Val600Glu mutation.32
Raf-1 has four activating phosphorylation sites that are Ras-inducible and confer full Raf activation: Ser338, Tyr340/341, Thr491, and Ser494. Mutation of these sites to phosphomimetic acidic residues results in constitutive activity independent of Ras.21 p21-activated Protein Kinase (PAK) and Sarcoma Viral Oncogene Homolog (Src) family members have been shown to phosphorylate Raf at Ser338 and Tyr340/341, respectively, and induce RAS activity.33,34,35 Other kinases that phosphorylate these activating residues have not been identified, and are the subject of considerable speculation. Phosphorylation also inhibits Raf. Raf-1 phosphorylated at Ser259 by Akt isoforms appears to create an auto-inhibitory conformation state maintained by 14-3-3 dimers, and mutation of this serine residue to an alanine residue restores the basal activity of Raf-1.36 Raf-1 can also undergo inhibitory phosphorylation at Ser289/296/301 by ERK, creating a negative feedback loop.37 The serine/threonine Protein Phosphatases 1 and 2a (PP1 and PP2A) target Ser259, thus promoting Raf-1 activation.38 Furthermore, Mammalian Ste20-like Kinase (MST2) indirectly inhibits Raf-1 phosphorylation at Ser259 by preventing PP2A catalytic subunit degradation.39 Interestingly, Raf inhibits MST2 activation and this inhibition does not require Raf kinase activity.40 Inhibition of PP1 and PP2A increases inactive 14-3-3/Raf-1 complex formation.
R&D Systems offers many tools to study your favorite signaling pathway
Simultaneously monitor multiple MAPK pathways
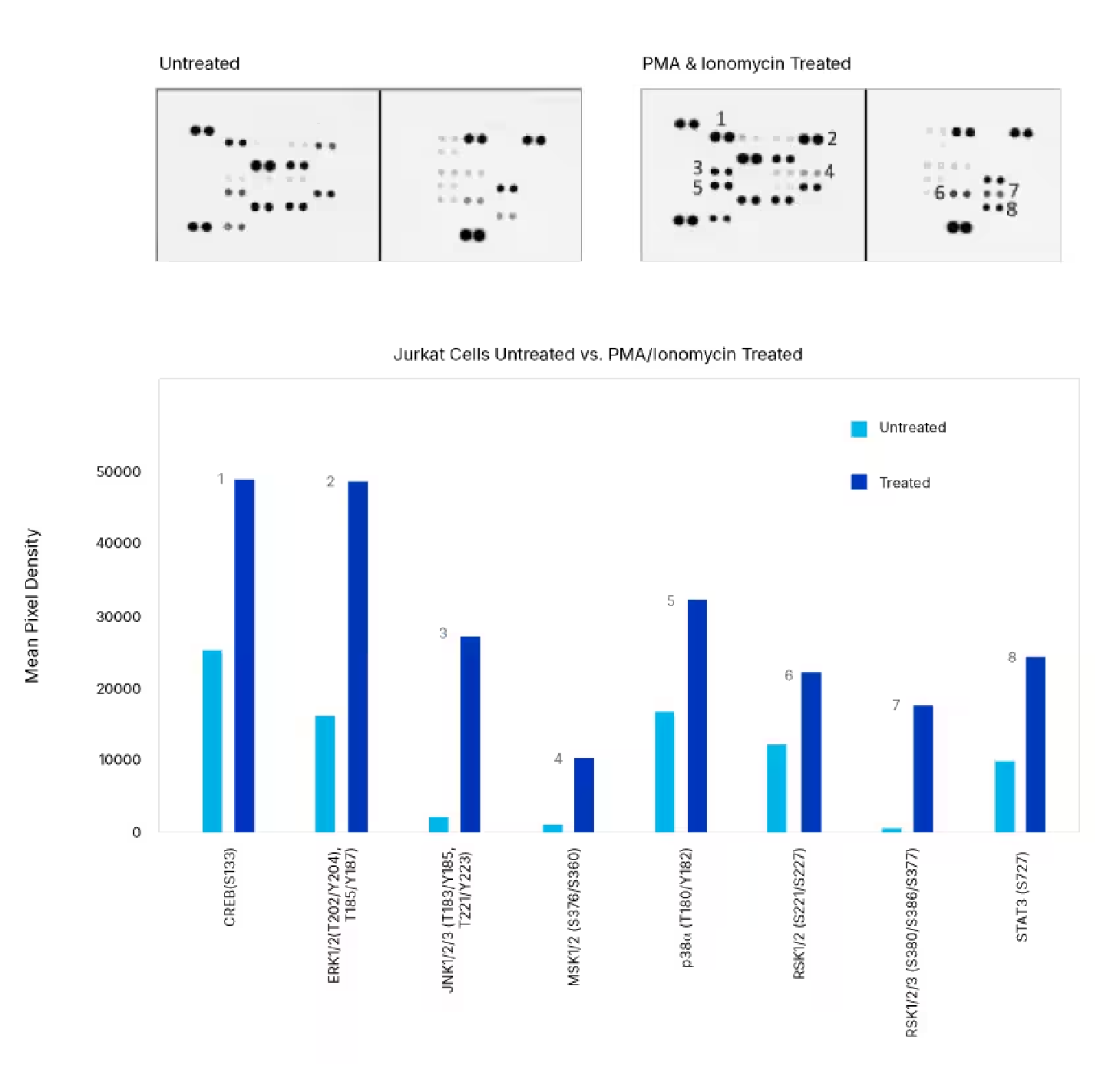
Detection of Human Protein Kinase Phosphorylation in PMA/ionomycin-treated Jurkat Cells. Jurkat human acute T cell leukemia cells were either left untreated or treated with 100 ng/mL PMA (Catalog # 1201) and 0.5 mM ionomycin for 20 minutes. The relative level of phosphorylation of 37 different kinases was simultaneously assessed using the Proteome Profiler™ Human Phospho-Kinase Antibody Array Kit (Catalog # ARY003C).
Verify phosphorylation with single analyte ELISAs
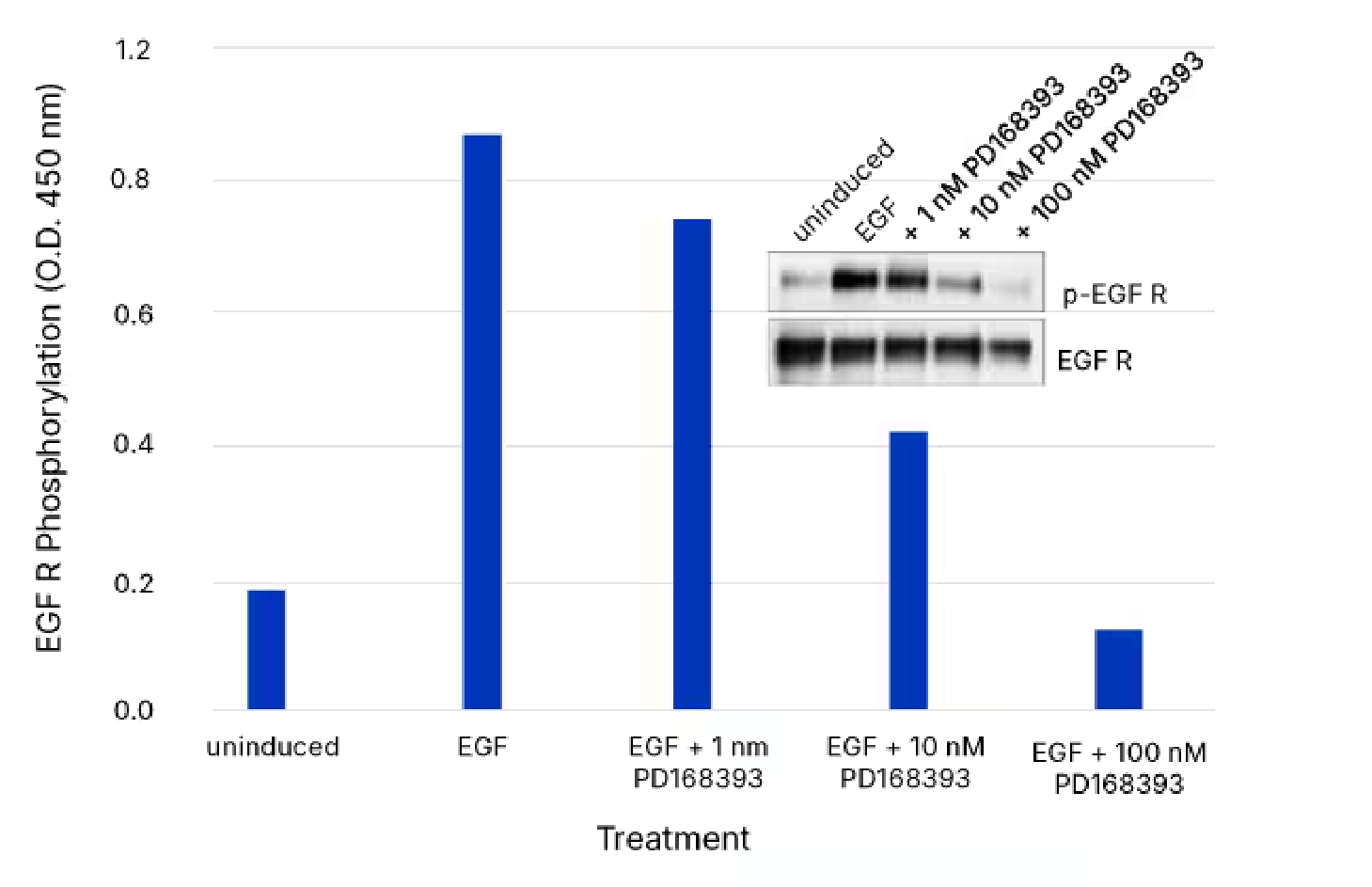
Use of the Human Phospho-EGF R DuoSet IC ELISA to Quantify Phospho-EGFR in Cells Pretreated with the inhibitor PD168393. The A431 human epidermoid carcinoma cell line was left untreated or treated with 25 ng/mL Recombinant Human EGF (Catalog # 236-EG) for 5 minutes, after either being treated without or with 1 nM, 10nM, or 100 nM PD168393. ELISA and IP-Western blot (inset) analyses were performed using 25 and 50 µg of lysate, respectively. IP-Western blots were done using an anti-EGFR monoclonal antibody and anti-mouse IgG agarose. Immunoblots were incubated with an HRP-conjugated Mouse Anti-Phospho-tyrosine Monoclonal Antibody (Catalog # HAM1676) to detect phosphorylated EGFR. Blots were stripped and total EGF R was detected using a Biotinylated Anti-EGF R Polyclonal Antibody (Catalog # BAF231). ELISA analysis of Phospho-EGFR was performed using the Human Phospho-EGF R DuoSet IC ELISA (Catalog # DYC1095B-2). The DuoSet IC ELISA results correlate well with the relative amount of phosphorylated EGFR detected by IP-Western blot.
Visualize cellular localization and expression
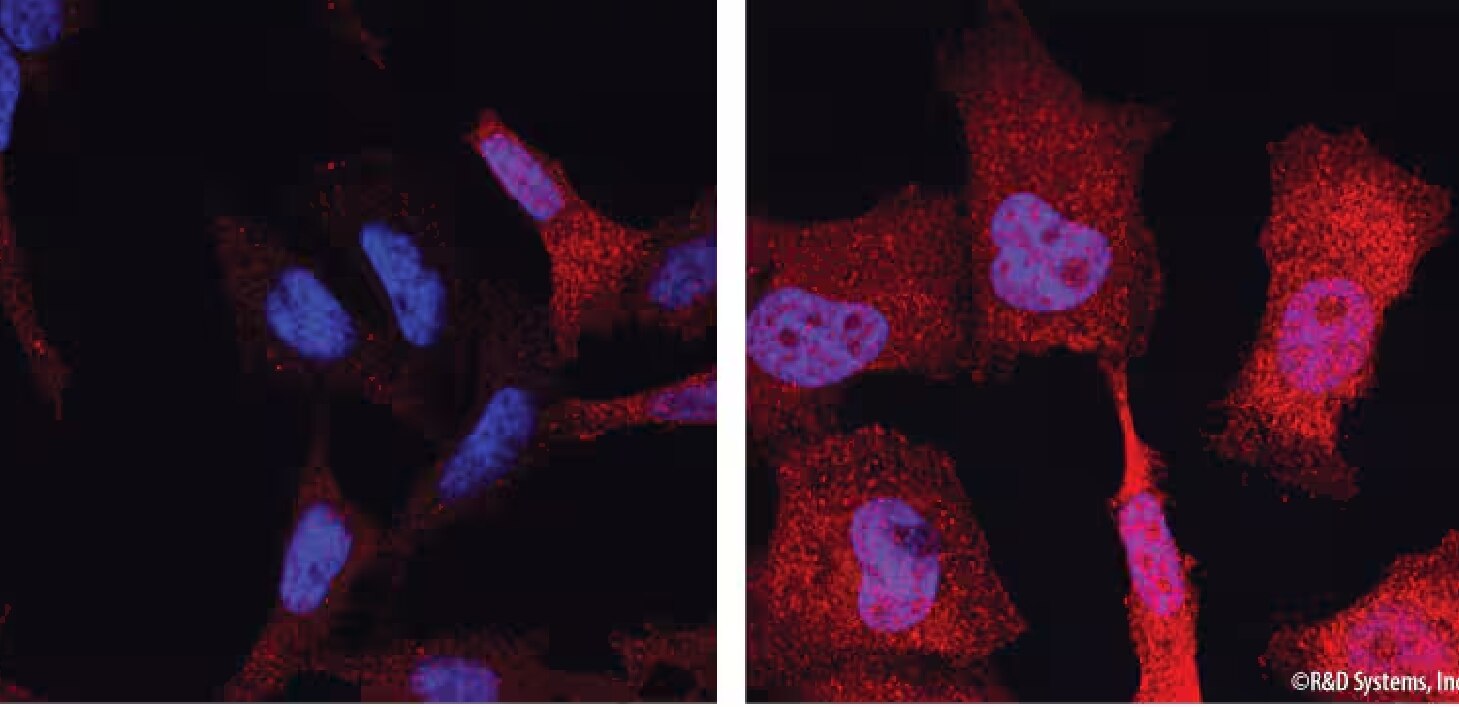
Detection of Phospho-ERK1/ERK2. ERK1 phosphorylated at T202/Y204 (ERK1) and T185/Y187 (ERK2) was detected in HeLa human cervical epithelial carcinoma cells that were either unstimulated (left panel) or stimulated (right panel) with PMA (Catalog # 1201) using a Rabbit Anti-Human/Mouse/Rat Phospho-ERK1 (T202/Y204)/ERK2 (T185/Y187) Monoclonal Antibody (Catalog # MAB1018). Cells were stained using a NorthernLights™ 557-conjugated Donkey Anti-Rabbit Secondary Antibody (red; Catalog # NL004). The nuclei were counterstained with DAPI (blue; Catalog # 5748).
Control your pathway with pharmacological inhibitors
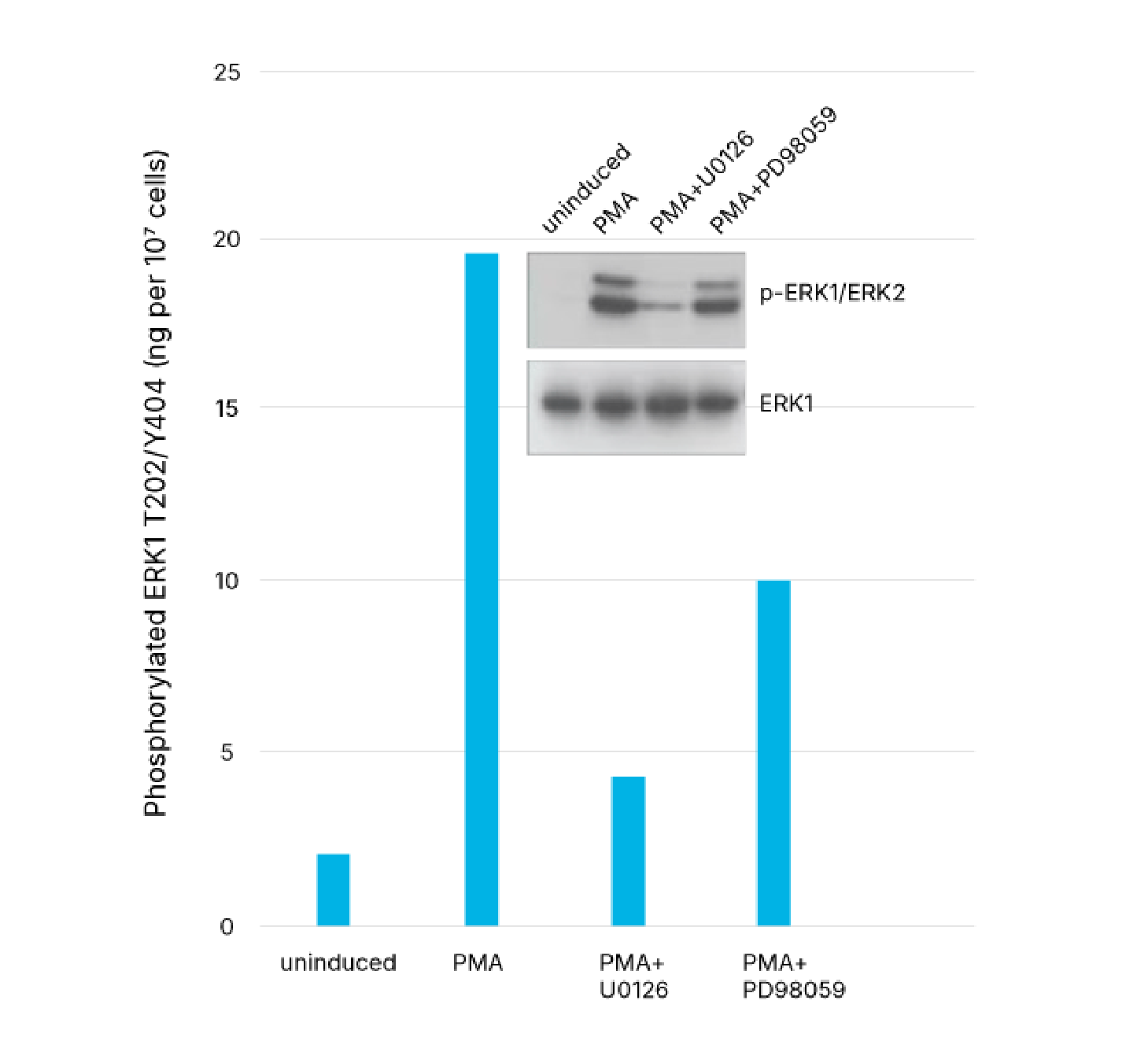
Quantification of Phosphorylated ERK1 in U0126- and PD 98059-treated Human HeLa Cells. HeLa human cervical epithelial carcinoma cells were untreated or treated with 200 nM PMA (Catalog # 1201) for 20 minutes, either with or without the MEK1/2 inhibitor, U0126 (50 mM; Catalog # 1144) or the MEK inhibitor, PD 98059 (50 mM; Catalog # 1213). Cells were lysed and ERK1 phosphorylated at T202/Y204 was quantified with the Human/Mouse/Rat Phospho-ERK1 (T202/Y204) DuoSet IC ELISA (Catalog # DYC1825-2). The same lysates were also immunoblotted (inset) with either Anti-Phospho-ERK1/ERK2 (p-ERK1/ERK2) or Anti-ERK1 polyclonal antibodies. The DuoSet IC ELISA results correlate well with the total amounts of phosphorylated ERK1 detected by Western blot. The blot with anti-ERK1 antibody indicates that the total levels of ERK1 remained constant during the various treatments. This DuoSet IC ELISA also quantifies phosphorylated ERK1 levels in mouse and rat cell lysates.
MEK
Phosphorylated Raf activates MAPK/ERK Kinase 1 (MEK1) and MEK2, also known as MAPK Kinase 1 (MKK1) and MKK2.41 Surprisingly, no naturally occurring oncoproteins derived from MEKs have been found, although expression of constitutively active forms transforms fibroblasts that produce tumors in nude mice.42Disruption of mouse mek1 is lethal in utero, with mutant embryos dying from defective placental vascularization, suggesting a role for MEK in angiogenesis.43MEK1 and MEK2, about 45 kDa each, share 80% sequence identity and contain a strong leucine-rich nuclear export signal (NES) at their N-termini.44,45Conservation of both forms throughout eukaryotic species suggests non-redundant functions, as does MEK1 gene disruption in mice.43 Additionally, MEK1 and MEK2 appear to be individually required for cell survival in vitro and MEK1 has been reported to downregulate MEK2-dependent ERK signaling, suggesting that MEK1 and MEK2 may not be completely interchangeable.46,47 Both MEKs are expressed ubiquitously in mammalian cells, although in mice MEK2 appears to be more highly expressed during development with MEK1 being more highly expressed in adult animals.48,49,50
Raf family activation of MEK1 and MEK2 occurs through phosphorylation of two serine residues at positions 217 and 221 found in the activation loop.51 MEKs can be partially activated by phosphorylation at either site, and substitution of these sites with acidic amino acids enhances basal activity.42 While Raf isoforms are enzymes of relatively low abundance, the high concentration of MEKs allows for amplification of signaling.52 Different Raf isoforms activate MEK1 and MEK2 differentially: A-Raf is a weak activator; B-Raf activates MEK1 preferentially; and Raf-1 efficiently activates both MEKs.53 Raf-1 contains two separate MEK binding sites, with substrate interaction greatly enhanced following phosphorylation of Raf-1 at Ser338.54 Two regulatory phosphorylation sites on MEK outside the activation loop either positively or negatively regulate the MAPKK. The first, at Ser298 and phosphorylated by PAK1, results in Raf-independent activation of MEK1.55 Conversely, in vivo phosphorylation by an unknown kinase at Ser212, a site conserved in all MAPKKs, sharply decreases MEK1 activity.56 Interestingly, quercitin, a red wine flavonoid, binds Ser212 and this also inhibits MEK1 activity.57
ERK
Also known as MAPK3 and MAPK1, the MAP kinases ERK1 and ERK2 are 44- and 42-kDa serine/threonine kinases, respectively, with 90% sequence identity in mammals. Initially isolated and cloned as kinases activated in response to insulin and Nerve Growth Factor (NGF), ERK1 and ERK2 are both expressed in most, if not all, mammalian tissues, with ERK2 levels generally higher than ERK1.58,59 Knock-out studies in mice demonstrate that either ERK may at least partially compensate for the other's loss, although ERK1 has been found to specifically regulate thymocyte maturation.2 Dual threonine and tyrosine residue phosphorylations activate both ERKs, at Thr202/Tyr204 for human ERK1 and Thr185/Tyr187 for human ERK2. Unlike MEK, significant ERK activation requires phosphorylation at both sites, with tyrosine residue phosphorylation preceding that of threonine.60 As with MEK, no in vivo mutations have been identified that activate ERK.
Because ERKs and other MAPKs require both threonine and tyrosine residue phosphorylation for full activity, Dual Specificity Phosphatases (DUSPs) that dephosphorylate both sites are uniquely positioned to regulate MAPK signal transduction cascades. At least 10 DUSPs, also termed MAPK Phosphatases (MKPs), have been identified in mammalian cells.61 DUSPs frequently associated with ERK inactivation include MKP3, MKP4, and Phosphatase of Activated Cells 1 (PAC1). MKP3, also termed DUSP6, is present in many tissues and most specific for ERKs versus other MAPKs. MKP4, expressed in kidney, placenta, and embryonic liver, strongly dephosphorylates ERKs but shows some reactivity toward Jun N-terminal Kinase (JNK) and p38 MAPK (p38) as well. The hematopoietically expressed PAC1 also shows limited reactivity with JNK and p38 in addition to ERKs, and is upregulated transcriptionally by p53.62 In addition to DUSPs, the phosphatases PP2A and Hematopoietic Protein-tyrosine Phosphatase (HePTP) have been implicated in ERK2 dephosphorylation at Thr185 and Tyr187, respectively.63 The finding that multiple phosphatases inactivate ERKs suggests that the duration and extent of ERK activation is controlled by the balanced activities of MEKs and these phosphatases.
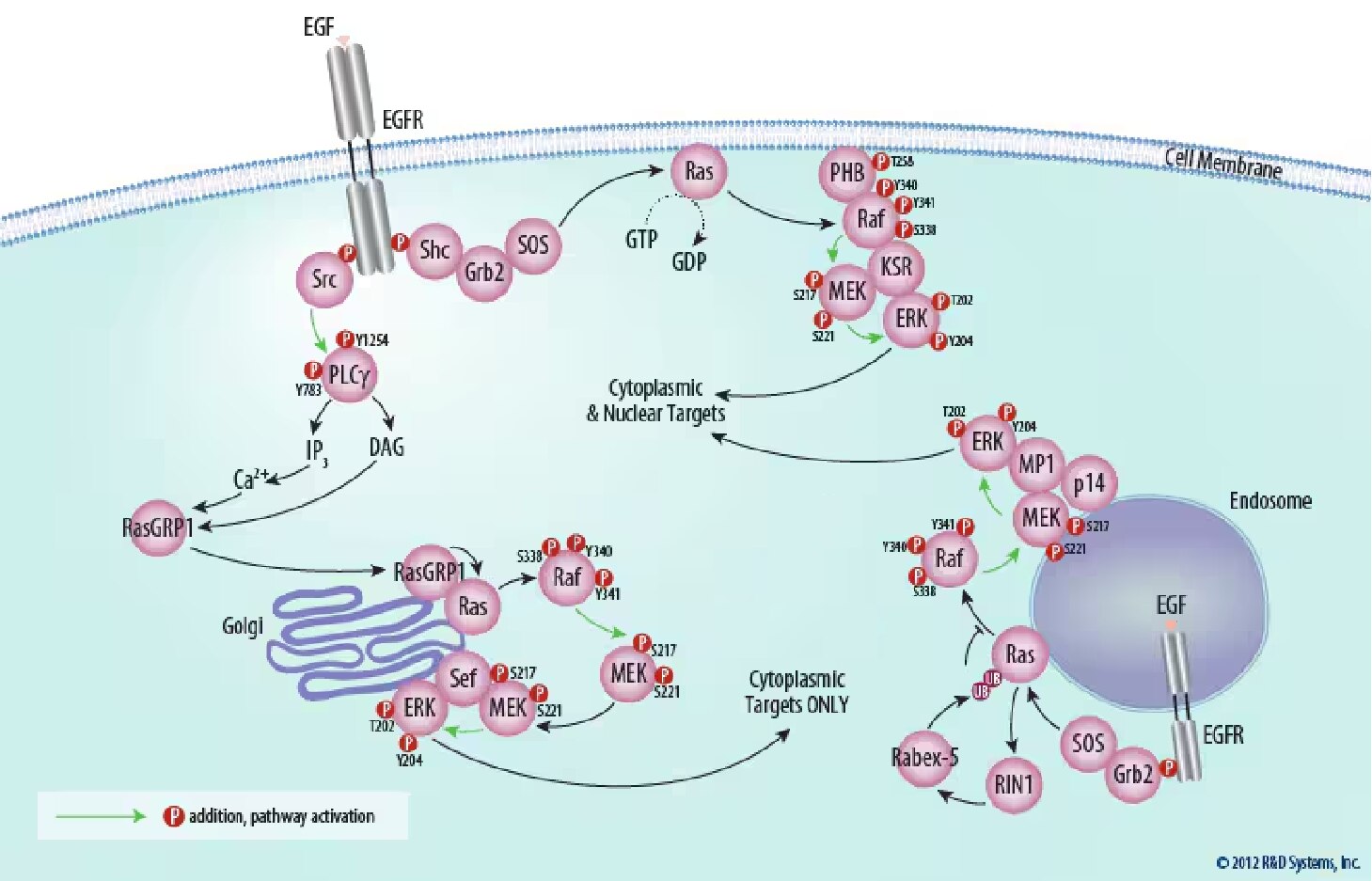
Figure 3. ERK Signaling is Compartmentalized Within the Cell. Distinct scaffold molecules are required for ERK activation at the plasma membrane (KSR), endosomes (MP1), and the golgi apparatus (Sef). ERK activated at the plasma membrane and endosomes is able to target both cytoplasmic and nuclear molecules. ERK activated at the golgi apparatus, however, is sequestered in the cytoplasm by Sef and is thus only able to target cytoplasmic molecules.
Studies have revealed scaffolding as a mechanism that helps the ERK cascade transduce signals with both high efficiency and specificity. Moreover, these scaffolds appear to be involved in spatial regulation of ERK signaling, as each scaffold restricts ERK signaling to a different subcellular compartment.64 First discovered in lower eukaryotes, experiments in mammals have focused on three scaffolding proteins in particular, Kinase Suppressor of Ras (KSR) and MEK Partner 1 (MP1), and Similar Expression to FGF Genes (Sef). KSR tethers Rafs, MEKs, and ERKs at the plasma membrane, facilitating ERK activation.65 Experiments with KSR-deficient mice indicate that KSR is not absolutely required, but enhances signaling from Ras.66 The scaffolding protein MP1 tethers MEK with ERK at endosomes, which is crucial for full ERK activation.67,68 Additionally, MP1-mediated endosomal ERK signaling is critical for a sustained ERK signal and tissue homeostasis in mice.69,70 Interestingly, Ras can be modified with K63-linked di-ubiquitin chains. Di-ubiquitinated Ras more stably associates with endosomes, but has a decreased ability to activate the Raf/MEK/ERK pathway.71Sef is a transmembrane scaffold for MEK and ERK that localizes to the Golgi apparatus. Importantly, Sef only binds activated MEK and inhibits the dissociation of the MEK-ERK complex, which blocks nuclear translocation of ERK, allowing specific activation of the cytoplasmic targets of ERK.72 Finally, ERK signaling from the Golgi apparatus appears to be functionally relevant, as it is necessary for positive thymocyte selection.73
Downstream from ERK
The ERKs are proline-directed protein kinases, phosphorylating proline-neighboring serine or threonine residues. Docking sites present on physiological substrates confer additional specificity.74 These docking interactions, through non-catalytic regions on ERK, team with scaffolding proteins to ensure signaling fidelity and enzymatic efficiency both to and from the MAPK. Downstream, activated ERK regulates growth factor-responsive targets in the cytosol and also translocates to the nucleus where it phosphorylates a number of transcription factors regulating gene expression (Figure 3).
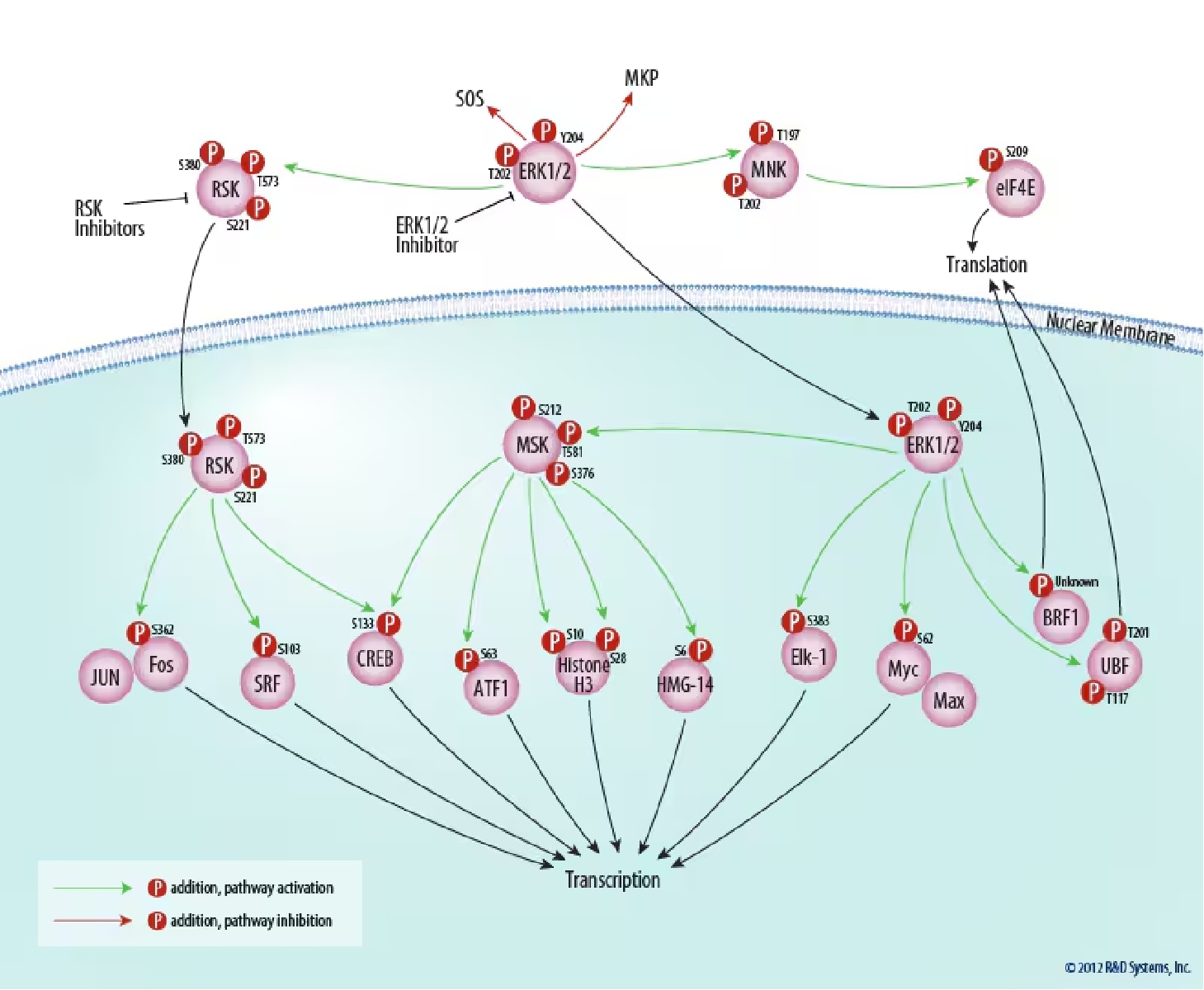
Figure 4. Downstream Targets of ERK Signaling. ERK1 and ERK2 positively regulate transcription directly and indirectly via phosphorylation of ribosomal protein S6 kinases (RSKs), mitogen- and stress-activated protein kinases (MSKs), and ternary complex factors (TCFs). Additionally, ERK1/2 indirectly regulate translation by inducing tRNA and rRNA synthesis. ERK1/2 also provide a negative feedback loop for the ERK signaling pathway via regulation of SOS and MKP. Tocris Biosciences provides inhibitors specific for RSK and ERK1/2.
Cytoplasmic Targets
Cytosolic substrates for ERK include several pathway components involved in ERK negative feedback regulation. Multiple residues on SOS are phosphorylated by ERK following growth factor stimulation.75,76 SOS phosphorylation destabilizes the SOS-GRB2 complex, eliminating SOS recruitment to the plasma membrane and interfering with Ras activation of the ERK pathway. Negative feedback by ERK also occurs through direct phosphorylation of the EGF receptor at Thr669, which inhibits its kinase activity.77,78 Finally, ERKs have also been demonstrated to negatively regulate themselves by phosphorylating MKPs, which reduces the degradation of these phosphatases through the Ubiquitin Proteasome System (UPS).79,80
MAPK-interacting Kinase 1 (MNK1) and MNK2 are cytosolic serine/threonine protein kinases initially discovered using two-hybrid screens for ERK-interacting proteins.81 Both ERK and p38, but not JNK, activate MNK by phosphorylation at Thr197 and Thr202. Active MNK1, and possibly MNK2, upregulates Eukaryotic Initiation Factor-4E (eIF-4E) in vitro, through phosphorylation at Ser209. Phosphorylation of eIF-4E at Ser209 is believed to strengthen this initiation factor's affinity for 7-methylguanosine cap structures, directing ribosomes to the 5' ends of mRNAs and enhancing translation efficiency. It has also been shown that MNK1 phosphorylation of eIF4E on Ser209 promotes tumorigenesis in mice, highlighting the importance of this modification.82
ERK1 and ERK2 regulate transcription indirectly by phosphorylating the 90 kDa Ribosomal Protein S6 Kinases (RSKs), a family of broadly expressed serine/threonine kinases activated in response to mitogenic stimuli, including growth factors and tumor-promoting phorbol esters.83 A highly conserved feature common to all RSK family members is the presence of tandem non-identical catalytic domains, involved in both exogenous phosphorylation and auto-activation.84 These domains are activated in a sequential manner by a series of phosphorylations following the binding of active ERK1 or ERK2 to an ERK docking site located at the extreme C-terminus of cytoplasmic RSK.85 On RSK1, these sequential phosphorylation sites include Thr573, Ser380 and Ser221. Active RSKs appear to play a major role in transcriptional regulation, translocating to the nucleus and phosphorylating such factors as the product of proto-oncogene c-fos at Ser362, serum response factor (SRF) at Ser103, and Cyclic AMP Response Element-binding Protein (CREB) at Ser133.86,87 Although RSK1 was initially purified and named based on its ability to phosphorylate the ribosomal protein S6 in vitro, this translational component is apparently the physiological substrate for the p70 S6 Kinases, and not the RSKs.88
Nuclear Targets
Upon phosphorylation, nuclear translocation of ERK1 and ERK2 is critical for both gene expression and DNA replication induced by growth factors.89 In the nucleus, ERK phosphorylates an array of targets, including transcription factors and a family of RSK-related kinases, the Mitogen- and Stress-activated Protein Kinases (MSKs).90 MSK1 and MSK2, activated by both ERK and p38, share the same tandem kinase structure as the RSKs, and also appear to be activated by sequential phosphorylation following MAPK docking. On MSK1, these sequential sites include Thr581, Ser376, and Ser212. MSKs phosphorylate and activate the Activator Protein 1 (AP-1) component Activating Transcription Factor 1 (ATF1) at Ser63, and may be more important in vivo than RSKs in CREB phosphorylation at the activating Ser133.91,92 Using knock-out mice, MSKs were also found to phosphorylate Histone H3 at Ser10 and Ser28, and the High-mobility-group protein HMG-14 at Ser6, facilitating the rapid induction of immediate early genes following mitogenic stimulation.93
Probably the best-characterized transcription factor substrates of ERKs are ternary complex factors (TCFs), including Ets-like Gene 1 (Elk-1), which is directly phosphorylated by ERK1 and ERK2 at multiple sites, including the activating Ser383.94 Upon complex formation with Serum Response Factor (SRF), phosphorylated TCFs transcriptionally activate the numerous mitogen-inducible genes regulated by serum response elements (SREs).95 The SREs regulating these genes are sufficient to confer inducibility to EGF, as well as to serum and phorbol esters. The TCFs SRF Accessory Protein 1 (Sap1) and Sap2 are also phosphorylated by ERK, and other E-twenty six (Ets) family members.96 Another direct target of ERK is the product of proto-oncogene v-Myc Myelocytomatosis Viral Oncogene Homolog (c-myc), a short-lived transcription factor involved in multiple aspects of growth control. Phosphorylation of Myc at Ser62 by ERK stabilizes Myc, allowing Myc to activate transcription as a heterodimeric partner with Myc Associated Factor X (Max).97 Conversely, phosphorylation of Myc at Thr58 by Glycogen Synthase Kinase 3 beta (GSK-3 beta), which is dependent on prior phosphorylation of Myc at Ser62, leads to proteasomal degradation of Myc via the F-box and WD Repeat Domain Containing 7 (FBW7) Ubiquitin ligase (E3).97,98
Finally, cellular growth and proliferation require protein synthesis, and the ERK pathway has been recently demonstrated to directly link growth factor signaling to ribosome biogenesis. Following serum induction, ERK phosphorylates the B-related Factor 1 (BRF1) subunit of RNA polymerase (pol) III-specific transcription factor (TFIIIB), both in vitro and in vivo, at an unknown site.99 As with MNK activation, phosphorylation of this pol III subunit enhances translational efficiency, inducing tRNA and 5S rRNA synthesis.Previous experiments demonstrated that ERK also upregulates synthesis of ribosomal RNA by pol I through phosphorylation of Upstream Binding Factor (UBF) at Thr117 and Thr201 following EGF treatment.100 Furthermore, ERK inhibits Tuberous Sclerosis 2 (TSC2) via phosphorylation at Ser664, which leads to TOR activation and increased translation.101
Conclusions
With aberrations in the ERK cascade implicated in a high proportion of human cancers, many emerging therapies target proteins in the pathway.102 Upstream, several drugs, including chimeric, humanized, and fully human monoclonal antibodies, along with small-molecule tyrosine kinase inhibitors (TKIs), are in clinical trials for EGF R family inhibition.103,104 Several Ras inhibitors have reached phase I to III clinical testing, including antisense oligonucleotides to limit expression and farnesyl transferase inhibitors to prevent membrane anchoring, but a lack of clinical success has reduced interest in Ras as a therapeutic target.105 However, the downstream kinases Raf and MEK 1/2 may be more promising targets.105 A number of small-molecule kinase inhibitors against Raf are in phase I to III clinical trials for the treatment of melanoma. 106,107,108 MEK 1/2 are attractive drug targets due to the potential minimizing of off-target effects since ERK 1/2 are the only known targets of MEK 1/2. 53,109 Despite the discontinued clinical use of some MEK inhibitors due to ineffectiveness and safety concerns, there have been promising phase I trial results with a second generation MEK inhibitor. 105,110,111,112 One area of concern for these treatments is patient resistance to the drugs. For example, 50% metastatic breast cancer patients have de novo resistance to Trastuzumab, a monoclonal antibody used to inhibit EGF R.113,114 Furthermore, patients in one study treated with a Raf inhibitor had an average of only 7 months of progression free survival.115 These examples highlight the continuing need for new drugs and targets and inhibition of the ERK pathway remains promising for cancer therapies.16,104
As these candidate therapies against ERK signaling components continue to undergo development and enter clinical trials, reagents that monitor inhibition of their targets are critical for future success. Unfortunately, it is frequently unclear in oncology research if a poor response is due to an inconsequential target or if the target was inhibited insufficiently. Providing a "proof of principle" tool for both the laboratory and the clinic, biomarker assays that correlate the extent of target suppression with the efficacy of treatment are needed.
References
- Seger, R. & E. Krebs (1995) FASEB J. 9:726.
- Pages, G. et al. (1999) Science 286:1374.
- Blume-Jensen, P. & T. Hunter (2001) Nature 411:355.
- Schlessinger, J. (2000) Cell 103:211.
- Downward, J. et al. (1984) Nature 307:521.
- Schlessinger, J. (2002) Cell 110:669.
- Zwick, E. et al. (1999) Trends Prot. Sci. 20:408.
- Levkowitz, G. et al. (1999) Mol. Cell 4:1029.
- Morandell, S. et al. (2008) Proteomics 8:4383.
- Li, N. et al. (1993) Nature 363:85.
- Rojas, M. et al. (1996) J. Biol. Chem. 271:27456.
- Pelicci, G. et al. (1992) Cell 70:93.
- Suenaga, A. et al. (2009) Biophys. J. 96:2278
- Shimizu, K. et al. (1983) Proc. Natl. Acad. Sci. USA 80:2112.
- Matallanas, D. & P. Crespo (2010) Curr. Opin. Mol. Ther. 12:674.
- Pyaleyeva-Gupta, Y. et al. (2011) Nat. Rev. Cancer 11:761.
- Avruch, J. et al. (1994) Trends Biochem. Sci. 19:279.
- Jelinek, T. et al. (1996) Mol. Cell. Biol. 16:1027.
- Rajalingam, K. et al. (2005) Nat. Cell Biol. 7:837
- Rapp, U. et al. (1983) Proc. Natl. Acad. Sci. USA 80:4218.
- Chong, H. et al. (2003) Cell. Signal. 15:463.
- Luckett, J.C. et al. (1996) Oncogene 12:1669.
- Provot, S. et al. (2008) Mol. Cell. Biol. 28:344.
- Galabova-Kovacs, G. et al. (2008) J. Cell. Biol. 180:947.
- Pritchard, C. et al. (1996) Curr. Biol. 6:614.
- Wojnowski, L. et al. (1997) Nat. Genet. 16:293.
- Mikula, M. et al. (2001) EMBO J. 20:1952.
- Fridman, M. et al. (2000) J. Biol. Chem. 275:30363.
- Chiu, C.F. et al. (2012) Oncogene Epub ahead of print.
- Yan, J. et al. (1998) J. Biol. Chem. 273:24052.
- Marais, R. et al. (1997) J. Biol. Chem. 272:4378.
- Tsao, H. et al. (2012) Genes Dev. 26:1131.
- Zang, M. et al. (2002) J. Biol. Chem. 277:4395.
- Edin, M. et al. (2005) Mol. Cell. Biol. 25: 4466.
- Alavi, A. et al. (2003) Science 301:94.
- Zimmermann, S.et al. (1999) Science 286:1741.
- Han, S. & K.E. Meier (2009) Cell. Signal. 21:793.
- Jaumot, M. & J. Hancock (2001) Oncogene 20:3949.
- Kilili, G.K. & J.M. Kyriakis (2010) J. Biol. Chem. 285:15076.
- O’Neill, E. et al. (2004) Science 306:2267.
- Zheng, C. & K. Guan (1993) J. Biol. Chem. 268:11435.
- Mansour, S. et al. (1994) Science 265:966.
- Giroux, S. et al. (1999) Curr. Biol. 9:369.
- Fukuda, M. et al. (1996) J. Biol. Chem. 271:20024.
- Fremin, C. & S. Meloche (2010) J. Hematol. Oncol. 3:8.
- Liu, X et al. (2004) Oncogene 23:765.
- Catalanotti, F. et al (2009) Nat. Struct. Mol. Biol. 16:294.
- Brott, B. et al. (1993) Cell Growth Differ. 4:921.
- Alessandrini, A. et al. (1997) Cell Growth Differ. 8:505.
- Voisin, L. et al. (2008) BMC Cancer 8:337.
- Alessi, D. et al. (1994) EMBO J. 13:1610.
- Huang, C. & J. Ferrell (1996) Proc. Natl. Acad. Sci. USA 93:10078.
- Kolch, W. (2000) Biochem. J. 351:289.
- Xiang, X. et al. (2002) J. Biol. Chem. 277:44996.
- Park, E et al. (2007) Cell Signal. 19:1488.
- Gopalbhai, K. et al. (2003) J. Biol. Chem. 278:8118.
- Lee, K. et al. (2008) Cancer Res. 68:946.
- Boulton, T. et al. (1990) Science 249:64.
- Boulton, T. et al. (1991) Cell 65:663.
- Ferrell, J. & R. Bhatt (1997) J. Biol. Chem. 272:19008.
- Caunt, C.J. & S.M. Keyse (2012) FEBS J. Epub ahead of print.
- Yin, Y. et al. (2003) Nature 422:527.
- Zhou, B. et al. (2002) J. Biol. Chem. 277:31818.
- Omerovic, J. & I.A. Poior (2009) FEBS J. 276:1817.
- Muller, J et al. (2001) Mol. Cell 8:983.
- Nguyen, A. et al. (2002) Mol. Cell. Biol. 22:3035.
- Wunderlich, W et al. (2001) J. Cell. Biol. 152:765.
- Teis, D. et al. (2002) Dev. Cell 3:803.
- Teis, D. et al. (2006) J. Cell Biol. 175:861.
- Taub, N. et al. (2007) Mol. Biol. Cell 18:4698.
- Jura, N. et al. (2006) Mol. Cell 21:679.
- Torii, S. et al. (2004) Dev. Cell 7:33.
- Daniels, M.A. et al. (2006) Nature 444:724.
- Tanoue, T. et al. (2000) Nature Cell Biol. 2:110.
- Dong, C. et al. (1996) J. Biol. Chem. 271:6328.
- Kamioka, Y. et al. (2010) J. Biol. Chem. 285:33540.
- Northwood, I. et al. (1991) J. Biol. Chem. 266:15266.
- Li, X. et al. (2008) Cell. Signal. 20:2145.
- Brondello, J.-M. et al. (1999) Science 286:2514.
- Peng, D. et al. (2010) Cell Cycle 9:4650.
- Waskiewicz, A. et al. (1997) EMBO J. 16:1909.
- Wendel, H et al. (2007) Genes Dev. 21:3232.
- Chen, R.-H. et al. (1991) Mol. Cell. Biol. 11:1861.
- Dalby, K. et al. (1998) J. Biol. Chem. 273:1496.
- Gavin, A.-C. & A. Nebreda (1999) Curr. Biol. 9:281.
- Chen, R.-H. et al. (1993) Proc. Natl. Acad. Sci. USA 90:10952.
- Xing, J. et al. (1996) Science 273:959.
- Chung, J. et al. (1992) Cell 69:1227.
- Brunet, A. et al. (1999) EMBO J. 18:664.
- Deak, M. et al. (1998) EMBO J. 17:4426.
- Gupta, P. & R. Prywes (2002) J. Biol. Chem. 277:50550.
- Wiggin, G. et al. (2002) Mol. Cell. Biol. 22:2871.
- Soloaga, A. et al. (2003) EMBO J. 22:2788.
- Marais, R. et al. (1993) Cell 73:381.
- Gille, H. et al. (1995) EMBO J. 14:951.
- Price, M. et al. (1995) EMBO J. 14:2589.
- Sears, R et al. (2000) Genes Dev. 14:2501.
- Welcker, M. et al. (2004) Proc. Natl. Acad. Sci. USA 101:9085.
- Felton-Edkins, Z. et al. (2003) EMBO J. 22:2422.
- Stefanovsky, V. et al. (2001) Mol. Cell 8:1063.
- Ma, L. et al. (2005) Cell 121:179.
- Santarpia, L. et al. (2012) Expert Opin. Ther. Targets 16:103.
- Eccles, S.A. (2011) Int. J. Dev. Biol. 55:685.
- Tsang, R.Y. & R.S. Finn (2012) Br. J. Cancer 106:6.
- Rusconi, P. et al. (2012) Curr. Med. Chem. 19:1164.
- Arkenau, H.T. et al. (2011) Br. J. Cancer 104:392.
- Chapman, P.B. et al. (2011) N. Engl. J. Med. 364:2507.
- Hauschild, A. et al. (2012) Lancet 380:358.
- Wei, F. et al. (2011) Curr. Med. Chem. 18:5476.
- Sebolt-Leopold, J.S. et al. (1999) Nat. Med. 5:810.
- Rinehart, J. et al. (2004) J. Clin. Oncol. 22:4456.
- Haura, E.B. et al. (2010) Clin. Cancer Res. 16:2450.
- Slamon, D.J. et al. (2001) N. Engl. J. Med. 344:783.
- Hurvitz, S.A. & R. Kakkar (2012) Ther. Adv. Med. Oncol. 4:235.
- Flaherty, K.T. et al. (2010) N. Engl. J. Med. 363:809.
Related Information
Apoptosis Intracellular Kinases
Intracellular Kinases in the Akt Pathway