R&D Systems Quality Control Western Blot Protocol
Introduction
Western blot is a research technique that utilizes antibodies to identify individual proteins within a cell or tissue lysate sample. Antibodies have the ability bind to highly specific sequences in a protein, known as an epitope. Due to this characteristic of antibodies, Western blot analysis can be used to identify and quantify a specific protein in a sample that can contain many different proteins. Western blot works by separating proteins in a sample based on their size using SDS-PAGE gel electrophoresis. Next, the proteins are transferred from the gel to a membrane by electrical current. After the proteins are transferred to the membrane, a primary antibody specific for the target of interest is added. Next, a secondary antibody with an attached enzyme is applied and finally a substrate that reacts with the secondary antibody-bound enzyme is added for detection and visualization.
R&D Systems Quality Control laboratories use this Western blot and immunostaining protocol to show that our antibodies are specific for the protein immunogen.
Protein samples are prepared with SDS and run under both reduced and non-reduced conditions on appropriate SDS-PAGE gel. The proteins are transferred to a PVDF membrane using a semi-dry transfer apparatus. For those proteins that have not been tested with natural samples, a protocol and troubleshooting guide is available for Western blot optimization. Please note that further optimization such as antibody concentration and buffers may be required.
Please read the following Western blot protocol in its entirety before beginning.

Contents
- Materials and Reagents Required
- Electrophoresis Protocol
- Transfer of Protein Protocol
- Immunostaining Protocol
- Data Examples
- Additional Western Blot Resources
Materials Required
- Semi-dry Electrophoretic Transfer Cell (Bio-Rad Catalog #1703940 or equivalent)
- Power supply 1-100 VDC (adj. current to 1 Amp)
- SDS-PAGE Gel
- PVDF Membrane, 0.45 µm pore size; cut to same size as gel (Millipore Catalog # IPVH304F0 or equivalent)
- Filter Paper, cut to same size as gel (Schleicher & Schuell 3MM or equivalent)
- Forceps, clean
- Plastic test tube
- Pipettes and pipette tips
Reagents Required
- Sample Buffer: 6% SDS, 0.25 M Tris, pH 6.8, 10% glycerol, 10 mM NaF and bromophenol blue with or without 20 mM dithiothreitol (DTT)
- Gel Running Buffer: 193 mM Glycine, 25 mM Tris (Base), 0.1% SDS
- 100% Methanol
- Anode Buffer I: 300 mM Tris, 20% methanol, pH 10.4
- Anode Buffer II: 25 mM Tris, 20% methanol, pH 10.4
- Cathode Buffer: 25 mM Tris, 20% methanol, 40 mM 6-aminocaproic acid, pH not adjusted
- Primary Antibody, polyclonal or monoclonal antibodies that have been validated for use in Western blots
- Secondary Antibody
- Streptavidin Alkaline Phosphatase (SA-AP) (Catalog # AR001) or equivalent
- Wash Solution (TTBS): 50 mM Tris, 0.5 M NaCl, 0.05% Tween 20, pH 7.4
- Blocking Buffer: 3% BSA (Fraction V, 3x crystallized), TTBS, 0.2% azide, pH 7.4
- Antibody Solution: 0.1% BSA in TTBS, pH 7.4
- Diluent Buffer: 1% BSA (Fraction V, 3x crystallized), TTBS, 0.2% azide, pH 7.4
- AP Substrate Buffer: 0.1 M Tris, 0.1 M NaCl, 5 mM MgCl2, pH 9.5
- Color Reagent A: 50 mg/mL nitro blue tetrazolium (NBT) in 70% dimethylformamide (DMF)
- Color Reagent B: 50 mg/mL bromochloroindolyl phosphate (BCIP) in 100% dimethylformamide (DMF)
Electrophoresis Protocol:
Purchase or prepare a SDS-PAGE gel that is appropriate for the estimated molecular weight (MW) of the protein of interest.
Note: Use 12% acrylamide gels for high MW proteins (>50 kDa), 15% gels for mid range MW proteins (15 - 50 kDa), and 20% gels for low MW proteins (<15 kDa).
- Treat samples by adding equal volumes of either 2X reducing or non-reducing Sample Buffer to protein sample solutions. Vortex gently to mix and then heat in boiling water bath for 5 minutes.
- Clamp the gel into an electrophoresis apparatus and add the Gel Running Buffer.
- Load samples onto gel.
- Connect electrophoresis unit to power supply.
- Start the electrophoresis process for the samples at 20 mA per gel. Run the gel at a constant current.
- Once the dye front has completely migrated into the running gel, increase current to 30 mA per gel.
- When the dye front reaches to about ½ the length of the running gel, increase current to 40 mA per gel.
- Electrophoresis is complete when the dye front migrates about 2 mm from the bottom of the gel.
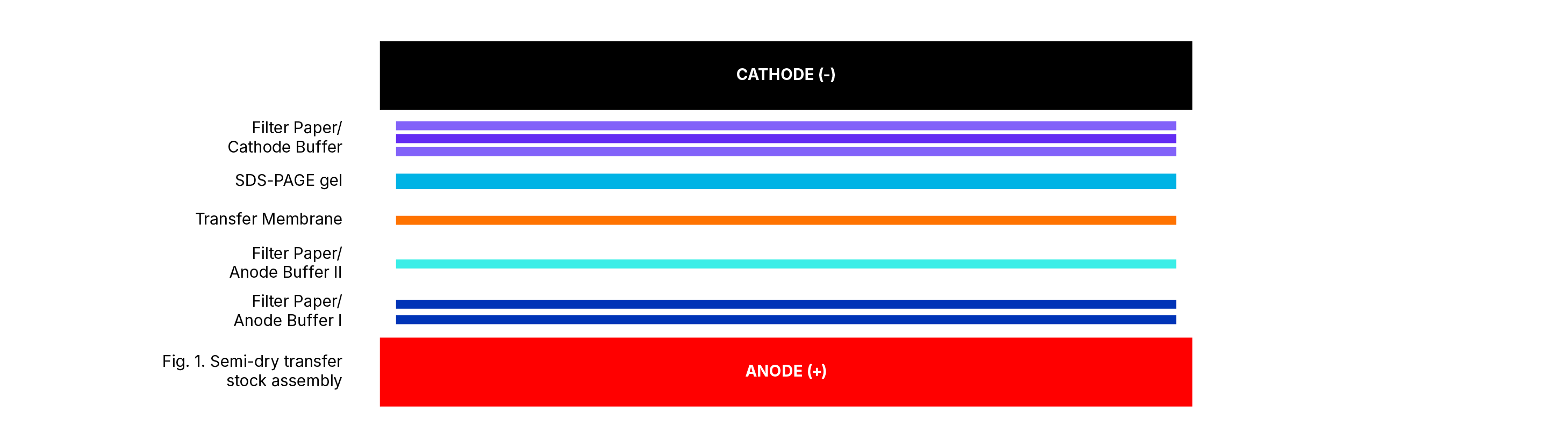
Transfer of Protein Protocol:
Note: Always wear gloves or use forceps when handling blotting membranes to avoid contamination with protein from fingers.
- Prepare the PVDF membrane by wetting the membrane with methanol for 15 seconds. The membrane should change from opaque to semi-transparent.
- Place membrane in de-ionized water and soak for 2 minutes.
- Place the membrane in Anode Buffer II for 5 minutes.
- Assemble Semi-dry transfer stack (Figure 1) by wetting two pieces of filter paper in Anode Buffer I and place on anode plate of Semi-dry Electrophoretic Transfer Cell. Avoid trapping air bubbles.
- Wet one piece of filter paper in Anode Buffer II and place on top of filter paper stack.
- Remove PVDF membrane from Anode Buffer II and place on top of filter paper stack.
- Place SDS-PAGE gel on top of PVDF membrane taking care not to trap air bubbles between gel and membrane.
- Wet three pieces of filter paper in Cathode Buffer and place on top of gel. Use a clean plastic test tube to roll out air bubbles.
- Place cathode plate of Semi-dry Electrophoretic Transfer Cell on top transfer stack.
- Connect high voltage cords to power supply. Apply a constant current of 1.9 - 2.5 mA per cm2 of gel area for 30 - 60 minutes. Appropriate transfer time must be determined empirically.
- After transfer is complete, turn off power supply and remove cathode plate of Semi-dry Electrophoretic Transfer Cell. Remove transfer membrane and cut lower right corner of membrane to mark orientation of the membrane.
- Discard the first two layers of filter paper and gel. Mark the bands of the molecular weight markers on the membrane with a ballpoint pen.
- Dry membrane in one of two ways.
- For fast drying, soak the membrane in 100% methanol for 10 seconds to drive out the water. Then, place the membrane on a piece of paper towel and leave on a lab bench for 15 minutes.
- For slower drying, place membrane on a piece of paper towel and leave on a lab bench for 2 hours or overnight.
- The dried membrane can be used immediately or stored in a dry container at 4°C for later use.
Immunostaining Protocol:
Note: Bring all the solutions and reagents to room temperature before use; otherwise the detection limit may be compromised.
Note: If membrane was dried and stored at 4°C, re-wet in 100% methanol for a few seconds. Discard methanol and rinse membrane in de-ionized water.
Block unoccupied protein binding sites on membrane by placing membrane in Blocking Buffer. Incubate on rocker/shaker at room temperature for 1-2 hours. Pour off the Blocking Buffer.
Note: if the primary antibody is conjugated, this step should be omitted and proceed directly to step 7
- Prepare the primary antibody in Antibody Solution at the recommended concentration listed on the datasheet. Add sufficient Antibody Solution with enough volume to cover the entire blot.
- Place container on a rocker and incubate at room temperature for one hour. Alternatively, the primary antibody incubation can be carried out at 4°C overnight to improve detection limit.
- Pour off Primary Antibody Solution and rinse membrane twice with de-ionized water. Then wash membrane twice in Wash Solution for 15 minutes each with shaking.
- Prepare secondary antibody in Antibody Solution as recommended on the datasheet. Add to blot container at volume sufficient to cover the entire membrane.
- Incubate on a rocker at room temperature for 30 minutes.
- Pour off secondary antibody solution and rinse membrane twice with de-ionized water. Then wash membrane twice in Wash Solution for 20 minutes each with shaking.
- Prepare the Streptavidin Alkaline Phosphatase (SA-AP) (Catalog # AR001) at 1:1000 dilution in Diluent Buffer. If using a different SA-AP, refer to manufacturer's instructions for dilution instructions. Add to blot container at a volume sufficient to cover the entire membrane.
- Incubate on a rocker at room temperature for 30 minutes.
- Pour off SA-AP solution and rinse membrane twice with de-ionized water. Then wash membrane twice in Wash Solution for 20 minutes each with shaking.
- Prepare the color solution by adding 132 mL of Color Reagent A stock solution to 20 mL of AP Substrate Buffer and mix well. Add 66 mL of Color Reagent B stock solution and mix well. Prepare substrate solution just before color development and use within 1 hour.
- For color development, add 20 mL of substrate solution and develop at room temperature with gentle shaking for 15 minutes or until desirable bands are developed.
- To stop color development, pour off the substrate solution and rinse the membrane with de-ionized water three times.
Data Examples

Detection of Human, Mouse, and Rat alpha Tubulin by Western Blot. Western blot shows lysates of HeLa human cervical epithelial carcinoma cell line, Jurkat human acute T cell leukemia cell line, NIH-3T3 mouse embryonic fibroblast cell line, Neuro-2A mouse neuroblastoma cell line, and Rat-2 rat embryonic fibroblast cell line. PVDF membrane was probed with 0.05 µg/mL of Mouse Anti-Human/Mouse/Rat a Tubulin Monoclonal Antibody (Catalog # MAB9344) followed by HRP-conjugated Anti-Mouse IgG Secondary Antibody (Catalog # HAF018). A specific band was detected for a Tubulin at approximately 52 kDa (as indicated). This experiment was conducted under reducing conditions and using Western Blot Buffer Group 1.

Detection of Human, Mouse, and Rat SOX2 by Western Blot. Western blot shows lysates of D3 mouse embryonic stem cell line, NTera-2 human testicular embryonic carcinoma cell line, F9 mouse teratocarcinoma stem cells, and rat cortical stem cells. PVDF membrane was probed with 1 µg/mL of Goat Anti-Human/Mouse/Rat SOX2 Antigen Affinity-purified Polyclonal Antibody (Catalog # AF2018) followed by HRP-conjugated Anti-Goat IgG Secondary Antibody (Catalog # HAF017). A specific band was detected for SOX2 at approximately 36 kDa (as indicated). This experiment was conducted under reducing conditions and using Western Blot Buffer Group 1.