Cytokines in Wound Healing
The complex interplay between multiple cytokines, cells and extracellular matrix is central to the initiation, progression and resolution of wounds.
Contents
- Introduction
- Platelet Activation and Cytokine Release
- Inflammation
- Re-epithelialization
- Granulation Tissue and Angiogenesis
- Matrix Production and Scar Formation
- Remodeling Phase
- Aberrant Healing
- References
Introduction
The response to injury is a phylogenetically primitive, yet essential innate host immune response for restoration of tissue integrity. Tissue disruption in higher vertebrates, unlike lower vertebrates, results not in tissue regeneration, but in a rapid repair process leading to a fibrotic scar. Wound healing, whether initiated by trauma, microbes or foreign materials, proceeds via an overlapping pattern of events including coagulation, inflammation, epithelialization, formation of granulation tissue, matrix and tissue remodeling. The process of repair is mediated in large part by interacting molecular signals, primarily cytokines, that motivate and orchestrate the manifold cellular activities which underscore inflammation and healing (Figure 1).
Response to injury is frequently modeled in the skin,1 but parallel coordinated and temporally regulated patterns of mediators and cellular events occur in most tissues subsequent to injury. The initial injury triggers coagulation and an acute local inflammatory response followed by mesenchymal cell recruitment, proliferation and matrix synthesis. Failure to resolve the inflammation can lead to chronic nonhealing wounds, whereas uncontrolled matrix accumulation, often involving aberrant cytokine pathways, leads to excess scarring and fibrotic sequelae. Continuing progress in deciphering the essential and complex role of cytokines in wound healing provides opportunities to explore pathways to inhibit/enhance appropriate cytokines to control or modulate pathologic healing.
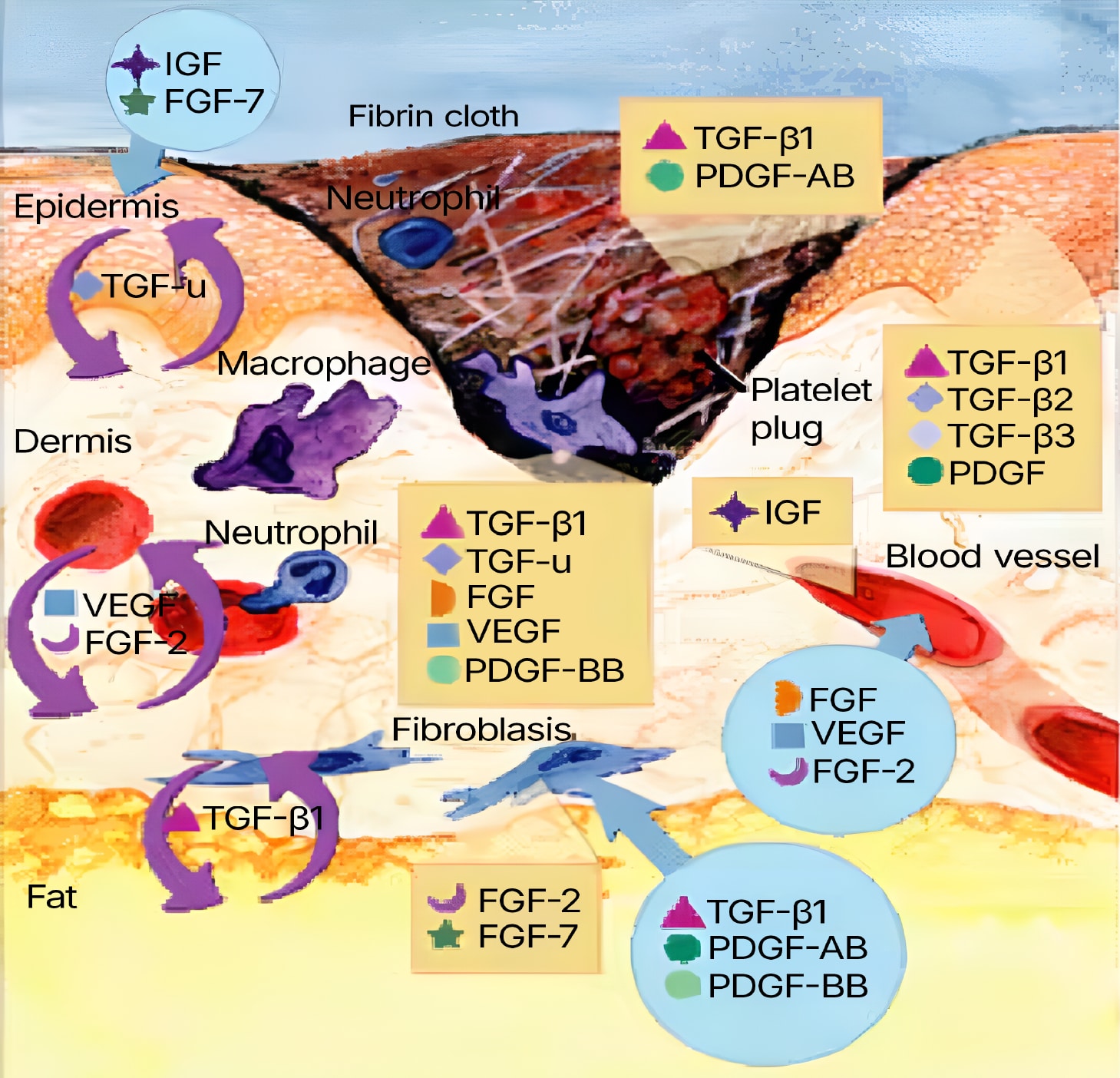
Figure 1. Wound healing is a complex process encompassing a number of overlapping phases, including inflammation, epithelialization, angiogenesis and matrix deposition. During inflammation, the formation of a blood clot re-establishes hemostasis and provides a provisional matrix for cell migration. Cytokines play an important role in the evolution of granulation tissue through recruitment of inflammatory leukocytes and stimulation of fibroblasts and epithelial cells. Adapted from Singer, A.J. & R.A. Clark (1999) New Engl. J. Med. 341:738.
Platelet Activation and Cytokine Release
Most types of injury damage blood vessels, and coagulation is a rapid-fire response to initiate hemostasis and protect the host from excessive blood loss. With the adhesion, aggregation and degranulation of circulating platelets within the forming fibrin clot, a plethora of mediators and cytokines are released (Table 1), including transforming growth factor beta (TGF-beta), platelet derived growth factor (PDGF), and vascular endothelial growth factor (VEGF), that influence tissue edema and initiate inflammation. VEGF, a vascular permeability factor, influences the extravasation of plasma proteins to create a temporary support structure upon which not only activated endothelial cells, but also leukocytes and epithelial cells subsequently migrate (see reference 2 for a review). Angiopoietin-1 (Ang-1), the ligand for Tie-2 receptors, is a natural antagonist for VEGF’s effects on permeability, a key regulatory checkpoint to avoid excessive plasma leakage.
Latent TGF-beta1, released in large quantities by degranulating platelets, is activated from its latent complex by proteolytic and non-proteolytic mechanisms3 to influence wound healing from the initial insult and clot formation to the final phase of matrix deposition and remodeling.4 Active TGF-beta1 elicits the rapid chemotaxis of neutrophils and monocytes to the wound site5 in a dose-dependent manner through cell surface TGF-beta serine/threonine type I and II receptors and engagement of a Smad3-dependent signal.6 Autocrine expression of TGF-beta1 by leukocytes and fibroblasts, in turn, induces these cells to generate additional cytokines including tumor necrosis factor alpha (TNF-a), interleukin 1 beta (IL-1 beta) and PDGF, as well as chemokines, as components of a cytokine cascade.7 Such factors act to perpetuate the inflammatory cell response, mediating recruitment and activation of neutrophils and monocytes. In response to TGF-beta and other cytokines, which engage their respective cell surface receptors, intracellular signaling pathways are mobilized to drive phenotypic and functional responses in target cell populations.8 Among the upstream signaling cascades engaged in acute tissue injury are NF-κB, Egr-1, Smads, and MAP kinases, which result in activation of many cognate target genes, including adhesion molecules, coagulation factors, cytokines and growth factors.8,9
Inflammation
Of the myriad of cytokines that have been investigated in terms of wound healing, TGF- beta 1 has undoubtedly the broadest effects. Despite the vast number of reports documenting the actions of TGF-beta in this context, both in vitro and in vivo, controversy remains as to its endogenous role. The paradoxical actions of TGF-beta are best appreciated in inflammation, where dependent upon the state of differentiation of the cell and the context of action, TGF-beta acts in a bi-directional manner.10 Moreover, this understanding of the nature of TGF-beta has led to the hypothesis that it may act as a therapeutic tool in some circumstances, but also a target for therapeutic intervention in others.10,11 Recent studies, in particular those utilizing genetically manipulated animal models, have highlighted the impact of TGF-beta on various aspects of wound healing, and surprisingly, not all of its effects are conducive to optimal healing. Intriguingly, mutations within the TGF-beta1 gene, or in the cell signaling intermediate Smad3, lead to normal or even accelerated cutaneous wound healing responses.6 The rate of healing of full-thickness wounds in Smad3 null mice was significantly greater than in their wild-type counterparts, associated with enhanced epithelialization and keratinocyte proliferation, and a markedly diminished inflammatory response. These observations have broad implications for understanding the role of TGF-beta in the endogenous wound healing response, in that an excess of TGF-beta may be a normal constituent of the response for rapid and optimal protection of the host. In the absence of infection, however, reduction of this overexuberant recruitment, inflammation and keratinocyte suppression may result in a more cosmetically acceptable scar. This knowledge may allow us to optimize the response by modulating selective cell pathways and to tailor therapy to specific cellular defects in pathological conditions such as chronic ulcers and fibrotic processes.
With the initial barrage of mediators, including TGF-beta, a chain reaction is set in motion, with recruitment, proliferation and activation of the cellular participants. Among the first cells to respond are the vascular endothelial cells, which not only respond to cytokines, but release them as well. Cytokine-induced enhancement of adhesion molecules (VCAM-1, ELAM-1, ICAM-1) on the endothelium provides the platform upon which circulating leukocytes expressing counter-adhesion molecules (integrins, selectins, Ig superfamily members) tether, slowing them down to sense the microenvironment and respond to chemotactic signals at the site of tissue injury.12 Adhesion molecule interactions between blood leukocytes and endothelium enables transmigration from inside to outside the vessel wall in response to multiple chemotactic signals. In addition to the powerful chemotactic activity of TGF-beta1 for neutrophils and monocytes,5,10 multiple chemokines are released to entice leukocytes into the site of tissue injury. Chemokines are represented by several families of related molecules based on the spatial location of the cysteine residues. Deletion of genes for chemokines leads to specific alterations in wound healing, underlying their role in this process (see references 13-15 for reviews).
Migrating through the provisional matrix (scaffolding) provided by the fibrin-enriched clot, leukocytes release proteases and engage in essential functions including phagocytosis of debris, microbes and degraded matrix components. Proteolytic activity is not constitutive, but transcriptionally driven by the cytokines, TGF-beta, IL-1beta and TNF-α, released from multiple cellular sources (Table 1). Neutrophil recruitment typically peaks around 24-48 hours post wounding, followed by an increasing representation of monocytes which are essential for optimal wound healing.16,17 Activation of these cells in the context of the wound microenvironment results in enhanced release of chemokines, recruitment of reinforcements, and amplification of the response, with the further release of cytokines, TNF-α, IL-1 and IL-6, that act as paracrine, autocrine and potentially, endocrine mediators of host defense. Antigen stimulation drives lymphocytic recruitment and activation, but at a delayed pace compared to the rapid acute response essential to maintain tissue integrity. Beyond the neutrophil, monocyte/macrophage and lymphocyte participants, mast cells have become increasingly recognized as active participants with increased numbers noted at sites of cutaneous injury.18 Mast cells respond to monocyte chemotactic protein (MCP-1) and TGF-beta1, -beta2 and -beta3, and within the lesion, release mediators (histamine, proteoglycans, proteases, platelet activating factor, arachidonate metabolites) and cytokines, including TGF-beta and IL-4 (Table 1). Once the inflammatory cells are activated, they become susceptible to TGF-beta1 mediated suppression to reverse the inflammatory process.7,10 Moreover, IL-4 may also dampen the inflammatory response as the inciting agent/trauma is dealt with and promote collagen synthesis during the repair phase.
Re-epithelialization
Clearance of debris, foreign agents, and/or infectious organisms promotes resolution of inflammation, apoptosis, and the ensuing repair response that encompasses overlapping events involved in granulation tissue, angiogenesis, and re-epithelialization. Within hours, epithelial cells begin to proliferate, migrate and cover the exposed area to restore the functional integrity of the tissue. Re-epithelialization is critical to optimal wound healing not only because of reformation of a cutaneous barrier, but because of its role in wound contraction. Immature keratinocytes produce matrix metalloproteases (MMPs) and plasmin to dissociate from the basement membrane and facilitate their migration across the open wound bed in response to chemoattractants. The migration of epithelial cells occurs independently of proliferation, and depends upon a number of possible processes including growth factors, loss of contact with adjacent cells, and guidance by active contact. TGF-beta1 stimulates migration of keratinocytes in vitro,6,19 possibly by integrin regulation and/or provisional matrix deposition.20 Behind the motile epidermal cells, basal cell keratinocyte proliferation is mediated by the local release of growth factors, with a parallel up-regulation of growth factor receptors including TNF-a, heparin-binding epidermal growth factor (EGF) and keratinocyte growth factor (KGF or FGF-7).21-23 Such growth factors are released not only by keratinocytes themselves, acting in an autocrine fashion, but also by mesenchymal cells and macrophages (Table 1), as paracrine mediators.24,25 Numerous animal models in which cytokine genes have been deleted or over-expressed have provided further evidence that such factors are involved in the process of epithelialization.23 TGF-beta1, and -beta2 are potent inhibitors of keratinocyte proliferation, with the Smad3 pathway implicated as the negative modulator.6 Since epithelialization is significantly accelerated in mice null for the Smad3 gene, with unchecked keratinocyte proliferation, but impaired migration in response to TGF-beta1, the implication is that the early proliferative event is critical to normal epithelialization.6 Once contact is established with opposing keratinocytes, mitosis and migration stop, and in the skin, the cells differentiate into a stratified squamous epithelium above a newly generated basement membrane. Other factors secreted by keratinocytes may exert paracrine effects on dermal fibroblasts and macrophages. One such factor is a keratinocyte-derived non-glycosylated protein termed secretory leukocyte protease inhibitor (SLPI), which inhibits elastase, mast cell chymase, NF-κB and TGF-β1 activation. In rodents, SLPI is a macrophage-derived cytokine with autocrine and paracrine activities,26, 27 but production by human macrophages has not yet been demonstrated. In mice, an absence of this mediator of innate host defense (SLPI null) is associated with aberrant healing.26
Granulation Tissue and Angiogenesis
Granulation tissue forms below the epithelium and is composed of inflammatory cells, fibroblasts and newly formed and forming vessels (Figure 2). This initial restructuring of the damaged tissue serves as a temporary barrier against the hostile external environment. Within granulation tissue, angiogenesis (i.e. the generation of new capillary blood vessels from pre-existing vasculature to provide nutrients and oxygen) is potentiated by hypoxia, nitric oxide (NO), VEGF and fibroblast growth factor 2 (FGF-2) (reviewed in references 2, 28) and by the chemokines, MCP-1 and macrophage inflammatory protein (MIP-1a).29 VEGF, released from wound epithelium and from the extracellular matrix by endothelial-derived proteases, stimulates endothelial cell proliferation and increases vascular permeability.2,30,31 VEGF may be transcriptionally up-regulated in response to NO, which also influences vasodilatation, an early step in angiogenesis. In a cyclic fashion, VEGF also drives nitric oxide synthase (NOS) in endothelial cells. Endothelial cells express high affinity receptors for VEGF, VEGF R1 (Flt-1) and VEGF R2 (Flk-1), and represent a primary target of this angiogenic and vascular permeability factor.31 Mice heterozygous for targeted inactivation of VEGF or homozygous for inactivation of its receptors are embryonically lethal, confirming the essentiality of VEGF in angiogenesis.32,33 Besides VEGF, FGFs transduce signals via four protein tyrosine kinase receptors34 to mediate key events involved in angiogenesis. FGFs recruit endothelial cells, and also direct their proliferation, differentiation and plasminogen activator synthesis. Clearly a multifactorial process, the cellular events underlying neovascularization are also impacted by TGF-beta1, EGF, TGF-a, endothelin 1, leptin, and indirectly, TNF-a and IL-1beta.
Of necessity, angiogenesis is a tightly controlled process. It is characterized not only by the presence of endogenous inducers, but also inhibitors which mediate a decline in the process as the granulation tissue, named for the granular appearance of the blood vessels in the wound, matures and scar remodeling continues. Among the identified endogenous inhibitors of re-vascularization are thrombospondin (TSP-1), IFN-?, IP-10, IL-12, IL-4 and the tissue inhibitors of MMPs (TIMPs), in addition to the recently recognized activities of angiostatin and endostatin (reviewed in reference 2). Since loss of angiogenic control may have negative consequences as evident in tumors, rheumatoid arthritis, and endometriosis, identification of potential endogenous and therapeutic modulators continues.

Figure 2. The remodeling phase (i.e. re-epithelialization and neovascularization) of wound healing is also cytokine-mediated. Degradation of fibrillar collagen and other matrix proteins is driven by serine proteases and MMPs under the control of the cytokine network. Granulation tissue forms below the epithelium and is composed of inflammatory cells, fibroblasts and newly formed and forming vessels. Adapted from Singer, A.J. & R.A. Clark (1999) New Engl. J. Med. 341: 738.
Matrix Production and Scar Formation
With the generation of new vasculature, matrix-generating cells move into the granulation tissue. These fibroblasts degrade the provisional matrix via MMPs and respond to cytokine/growth factors by proliferating and synthesizing new extracellular matrix (ECM) to replace the injured tissue with a connective tissue scar. Although the stage is being set for tissue repair from the beginning (provisional matrix, platelet release of PDGF and TGF-beta, cytokine reservoir), fibroblasts migrate into the wound and matrix synthesis begins in earnest within a couple of days, continuing for several weeks to months. TGF-beta contributes to the fibrotic process by recruiting fibroblasts and stimulating their synthesis of collagens I, III, and V, proteoglycans, fibronectin and other ECM components.4,35 TGF-beta concurrently inhibits proteases while enhancing protease inhibitors, favoring matrix accumulation. In vivo studies have confirmed that exogenous TGF-beta1 increases granulation tissue, collagen formation, and wound tensile strength when applied locally or given systemically in animal models. Increased levels of TGF-beta are routinely associated with both normal reparative processes, as well as fibropathology. In Smad3 null mouse wounds, matrix deposition (fibronectin) could be restored by exogenous TGF-beta, implying a Smad3-independent pathway, whereas collagen deposition was not restored, suggesting a dichotomous Smad3-dependent regulation.6 The progressive increase in TGF-beta3 over time and its association with scarless fetal healing have implicated this member of the TGF-beta family in the cessation of matrix deposition.36 Other members of the TGF-beta superfamily may also contribute to the wound healing response. Activin A when over-expressed in basal keratinocytes stimulates mesenchymal matrix deposition,37 whereas BMP-6 over-expression inhibits epithelial proliferation.38
PDGF, released at the outset by degranulating platelets, represents a family of cytokines consisting of two polypeptide chains (A and B) which form the dimers PDGF-AA, AB and B.39 In addition to platelets, PDGF is released by activated macrophages, endothelial cells, fibroblasts and smooth muscle cells (Table 1) and is a major player in regulating fibroblast and smooth muscle cell recruitment and proliferation through PDGF specific receptor-ligand interactions.40 Beyond its role in fibroblast migration and matrix deposition, PDGF-A and -B also up-regulate protease production, in contrast to the anti-protease activity of TGF-beta.41,42 PDGF represents the only FDA approved cytokine/growth factor for the clinical enhancement of delayed wound healing. Also central to repair are the FGFs, which signal mitogenesis and chemotaxis,34 underlying granulation tissue formation, and the production of MMPs.43 FGF-1 (acidic FGF) and FGF-2 (basic FGF) have been the most intensely studied, but the additional members of this family may also support tissue repair and/or have clinical application.44 The role of FGF-2 has been confirmed in the FGF-2 null mouse which shows not only retarded epithelialization but also reduced collagen production.45
With many overlapping functional properties with FGFs, epidermal growth factor (EGF) orchestrates recruitment and growth of fibroblasts and epithelial cells in the evolution of granulation tissue. EGF and TGF-a, which share sequence homology, enhance epidermal regeneration and tensile strength in experimental models of chronic wounds.46 TNF-a and IL-1beta, key mediators of the inflammatory process, also contribute to the reparative phase either directly by influencing endothelial and fibroblast functions or indirectly, by inducing additional cytokines and growth factors. IL-6 has also been shown to be crucial to epithelialization and influences granulation tissue formation, as shown in the wound healing studies of mice null for the IL-6 gene.47 As repair progresses, fibroblasts display increased expression levels of adhesion molecules and assume a myofibroblast phenotype, mediated in part by TGF-beta and PDGF-A and -B, to facilitate wound contraction.48
Remodeling Phase
The remodeling phase, during which collagen is synthesized, degraded and dramatically reorganized (as it is stabilized via molecular crosslinking into a scar), is also cytokine-mediated. Although repaired tissue seldom achieves its original strength, it provides an acceptable alternative. Degradation of fibrillar collagen and other matrix proteins is driven by serine proteases and MMPs under the control of the cytokine network. MMPs not only degrade matrix components, but also function as regulatory molecules by driving enzyme cascades and processing cytokines, matrix and adhesion molecules to generate biologically active fragments. TIMPs provide a natural counterbalance to the MMPs and disruption of this orderly balance can lead to excess or insufficient matrix degradation and ensuing tissue pathology.49 Similarly, there exists a naturally occurring inhibitor of elastase and other serine proteases (i.e. SLPI).26,27 The coordinated regulation of enzymes and their inhibitors ensures tight control of local proteolytic activity. In physiologic circumstances, these molecular brakes limit tissue degradation and facilitate accumulation of matrix and repair.
Aberrant Healing
Rapid clearance of the inciting agent and resolution of inflammation during healing minimizes scar formation, whereas persistence of the primary insult results in continued inflammation and chronic attempts at healing. Prolonged inflammation and proteolytic activity prevent healing as evident in ulcerative lesions. On the other hand, continued fibrosis in the skin leads to scarring and potentially, disfigurement, whereas progressive deposition of matrix in internal organs such as lungs, liver, kidney or brain compromises not only their structure, but also function, causing disease and death. Inhibitors of TGF-beta (e.g. antibodies, decorin, Smad 7, antisense oligonucleotides)50-52 reduce scarring, as does local administration of exogenous TGF-beta336 or systemic delivery of TGF-beta1.53 IFN-γ is a natural antagonist of fibrogenesis through its ability to inhibit fibroblast proliferation and matrix production and has been shown to have clinical efficacy.54,55 IL-10 may be considered anti-fibrotic via its anti-inflammatory activities,56 as are inhibitors of TNF-a.57
Wound healing is a complex process encompassing a number of overlapping phases, including inflammation, epithelialization, angiogenesis and matrix deposition. Ultimately these processes are resolved or dampened leading to a mature wound and macroscopic scar formation. Although inflammation and repair mostly occur along a proscribed course, the sensitivity of the process is underscored by the consequences of disruption of the balance of regulatory cytokines. Consequently, cytokines, which are central to this constellation of events, have become targets for therapeutic intervention to modulate the wound healing process. Depending on the cytokine and its role, it may be appropriate to either enhance (recombinant cytokine, gene transfer) or inhibit (cytokine or receptor antibodies, soluble receptors, signal transduction inhibitors, antisense) the cytokine to achieve the desired outcome.
References
- Singer, A.J. & R.A. Clark (1999) New Engl. J. Med. 341:738.
- Liekens, S. et al. (2001) Biochem. Pharmacol. 61:253.
- Khalil, N. (1999) Microbes Infect. 1:1255.
- Wahl, S.M. (1999) “Transforming growth factor beta.” in Inflammation: Basic Principles and Clinical Correlates, Third Edition, J. Gallin and R. Snyderman, eds., Lippincott-Raven Publishers, Philadelphia, pp. 883-892.
- Wahl, S.M. et al. (1987) Proc. Natl. Acad. Sci. USA 84:5788.
- Ashcroft, G.S. et al. (1999) Nat. Cell Biol. 1:260.
- McCartney-Francis, N.L. & S.M. Wahl (2001) “TGF-beta and macrophages in the rise and fall of inflammation.” in TGF-beta and Related Cytokines in Inflammation, Breit, S.N. and S.M. Wahl, ed., Birkhauser, Basel, pp. 65-90.
- Heldin, C.H. et al. (2001) “Signal transduction mechanisms for members of the TGF-beta family.” in TGF-beta and Related Cytokines in Inflammation, Breit, S.N. and S.M. Wahl ed., Birkhauser, Basel, pp. 11-40.
- Braddock, M. (2001) Ann. Med. 33:313.
- Wahl, S.M. (1994) J. Exp. Med. 180:1587.
- Chen, W. & S.M. Wahl (1999) Microbes Infect. 1:1367.
- Sundy, J.S. & B.F. Haynes (2000) Curr. Rheumatol. Rep. 2:402.
- Gillitzer, R. & M. Goebeler (2001) J. Leukoc. Biol. 69:513.
- Cacalano, G. et al. (1994) Science 265:682.
- Morales, J. et al. (1999) Proc. Natl. Acad. Sci. USA 96:14470.
- Leibovich, S.J. & R. Ross (1975) Am. J. Pathol. 78:71.
- Clarke, R.A.F. (1996) “Wound repair: overview and general considerations” in The Molecular and Cellular Biology of Wound Repair, Clark, R.A.F. ed., Plenum, New York, pp. 3-50.
- Huttunen, M. et al. (2000) Exp. Dermatol. 4:258.
- Hebda, P.A. (1998) J. Invest. Derm. 91:440.
- Wikner, N.E. et al. (1998) J. Invest. Derm. 91:207.
- Barrandon, Y. & H. Green (1987) Cell 50:1131.
- Higashiyama, S. et al. (1991) Science 251:936.
- Werner, S. et al. (1994) J. Invest. Derm. 103:469.
- Coffey, Jr., R.J. et al. (1987) Nature 328:817.
- Werner, S. et al. (1992) Proc. Natl. Acad. Sci. USA 89:6896.
- Ashcroft, G.S. et al. (2000) Nat. Med. 6:1147.
- Song, X.Y. et al. (1999) J. Exp. Med. 190:535.
- Conway, E.M. et al. (2001) Cardiovascul. Res. 49:507.
- Belperio, J.A. et al. (2000) J. Leukoc. Biol. 68:1.
- Brown, L.F. et al. (1992) J. Exp. Med. 176:1375.
- Ferrara, N. (1999) Curr. Top. Microbiol. Immunol. 237:1.
- Ferrara, N. et al. (1996) Nature 380:439.
- Shibuya, M. (2001) Int. J. Biochem. Cell Biol. 33:409.
- Ornitz, D.M. et al. (1996) J. Biol. Chem. 271:15292.
- Branton, M.H. & J.B. Kopp (1999) Microbes Infect. 1:1349.
- Niesler, C.U. & M.W.J. Ferguson. (2001) “TGF-beta superfamily cytokines in wound healing” in TGF-beta and Related Cytokines in Inflammation (Breit, S.N. and S.M. Wahl, ed., Birkhauser, Basel, pp. 173-198.
- Munz, B. et al. (1999) EMBO J. 18:5205.
- Blessing, M. et al. (1996) J. Cell Biol. 135:227.
- Ross, R. et al. (1990) Philos. Trans. R. Soc. Lond. B. Biol. Sci. 327:155.
- Claesson-Welsh, L. (1996) Int. J. Biochem. Cell Biol. 28:373.
- Circolo, A. et al. (1991) J. Biol. Chem. 266:12283.
- Laiho, M. et al. (1987) J. Biol. Chem. 262:17467.
- Powers, C.J. et al. (2000) Endocr. Relat. Cancer 7:165.
- Payne, W.G. et al. (2001) Am. J. Surg. 181:81.
- Ortega, S. et al. (1998) Proc. Natl. Acad. Sci. USA 95:5672.
- Andresen, J.L. & N. Ehlers (1998) Curr. Eye Res. 17:79.
- Gallucci, R.M. et al. (2000) FASEB J. 14:2525.
- Grinnell, F. (1994) J. Cell Biol. 124:401.
- Birkedal-Hansen, H. (1995) Curr. Opin. Cell Biol. 7:728.
- Wahl, S.M. et al. (1993) J. Exp. Med. 177:225.
- Border, W.A. & N.A. Noble (1998) Kidney Int. 54:1390.
- Nakao, A. et al. (1999) J. Clin. Invest. 104:5.
- Song, X. et al. (1999) J. Immunol. 163:4020.
- Ghosh, A.K. et al. (2001) J. Biol. Chem. 276(14):11041.
- Duncan, M.R. et al. (1985) J. Exp. Med. 162(2):516.
- Akdis, C.A. (2001) Immunology 103(2):131.
- Wahl, S.M. et al. “Cytokine modulation in the therapy of hepatic immunopathology and fibrosis” in Cytokines in Liver Injury and Repair. Kluwer Academic Publishers, Lancaster. (2002)