Publication Spotlight: Disruption of IL-11 Signaling Inhibits Cellular Senescence and Extends Healthy Aging in Mice
Aging and Cellular Senescence
Aging is a gradual, irreversible process associated with a progressive loss of tissue and organ functions, chronic, low-grade inflammation, and an increased risk of age-related diseases.1 These changes are in part attributed to an accumulation of senescent cells that have undergone a stress-induced program of permanent cell cycle arrest and display a pro-inflammatory, senescence-associated secretory phenotype (SASP).
While acute cellular senescence during embryonic development and wound healing is beneficial as a short-term process that controls cell growth and tissue organization, chronic senescence that occurs during aging is more deleterious. During acute senescence, inflammatory cytokines secreted by senescent cells promote immune cell recruitment, leading to removal of the senescent cells and restoration of tissue homeostasis. In contrast, age-related chronic senescence stems from both an increase in the production of senescent cells and a decline in the ability of the immune system to clear these cells known as immunosenescence.2 The resulting accumulation of senescent cells is directly linked to chronic inflammation or “inflammaging”, which can lead to age-related organ damage and diseases such as cancer, atherosclerosis, type 2 diabetes, and neurodegenerative disorders.3 The connection between senescence, aging, and age-related disease was initially demonstrated by Baker, et al., who showed that the removal of senescent cells in mice could extend their lifespan, prevent age-related organ deterioration, and delay tumorigenesis.4
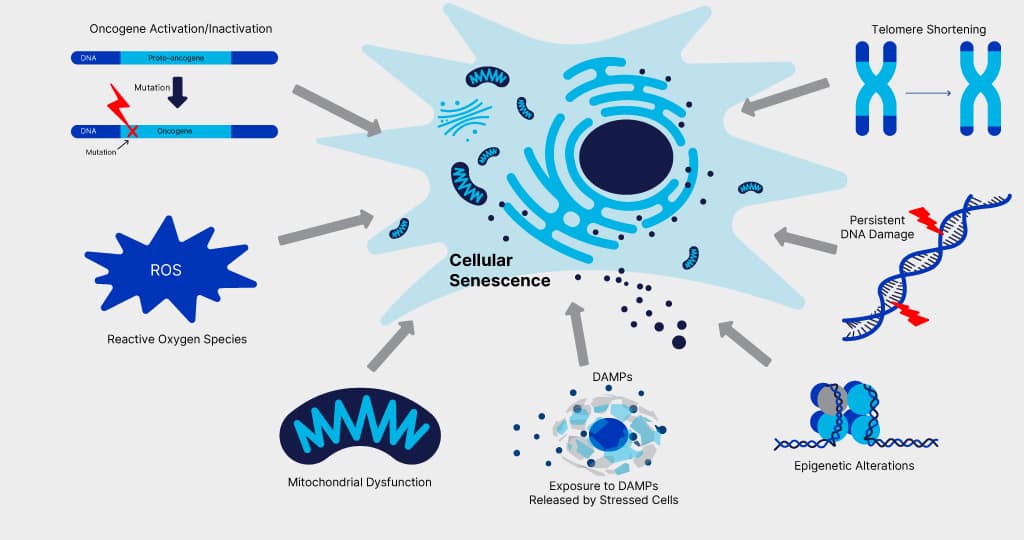
Figure 1. Triggers of Cellular Senescence. Cellular senescence can be triggered in response to a variety of stress signals including oncogene activation/inactivation, high levels of reactive oxygen species (ROS), mitochondrial dysfunction, exposure to damage-associated molecular patterns (DAMPs) released by damaged cells, epigenetic alterations, persistent DNA damage, and telomere shortening.
IL-11 Signaling is Linked to Senescence and Inflammaging
A pioneering study published earlier this year in Nature has now identified IL-11 as a senescence-associated cytokine that regulates inflammaging, lifespan, and the development of age-related diseases in mice.5 IL-11 is a pro-inflammatory and pro-fibrotic member of the IL-6 cytokine family that is up-regulated with age.6, 7 It signals through a receptor complex consisting of IL-11 Rα and gp130 to activate multiple intracellular signaling pathways, including the Jak-STAT, PI3K-Akt-mTORC1, and Ras-MEK-ERK pathways.
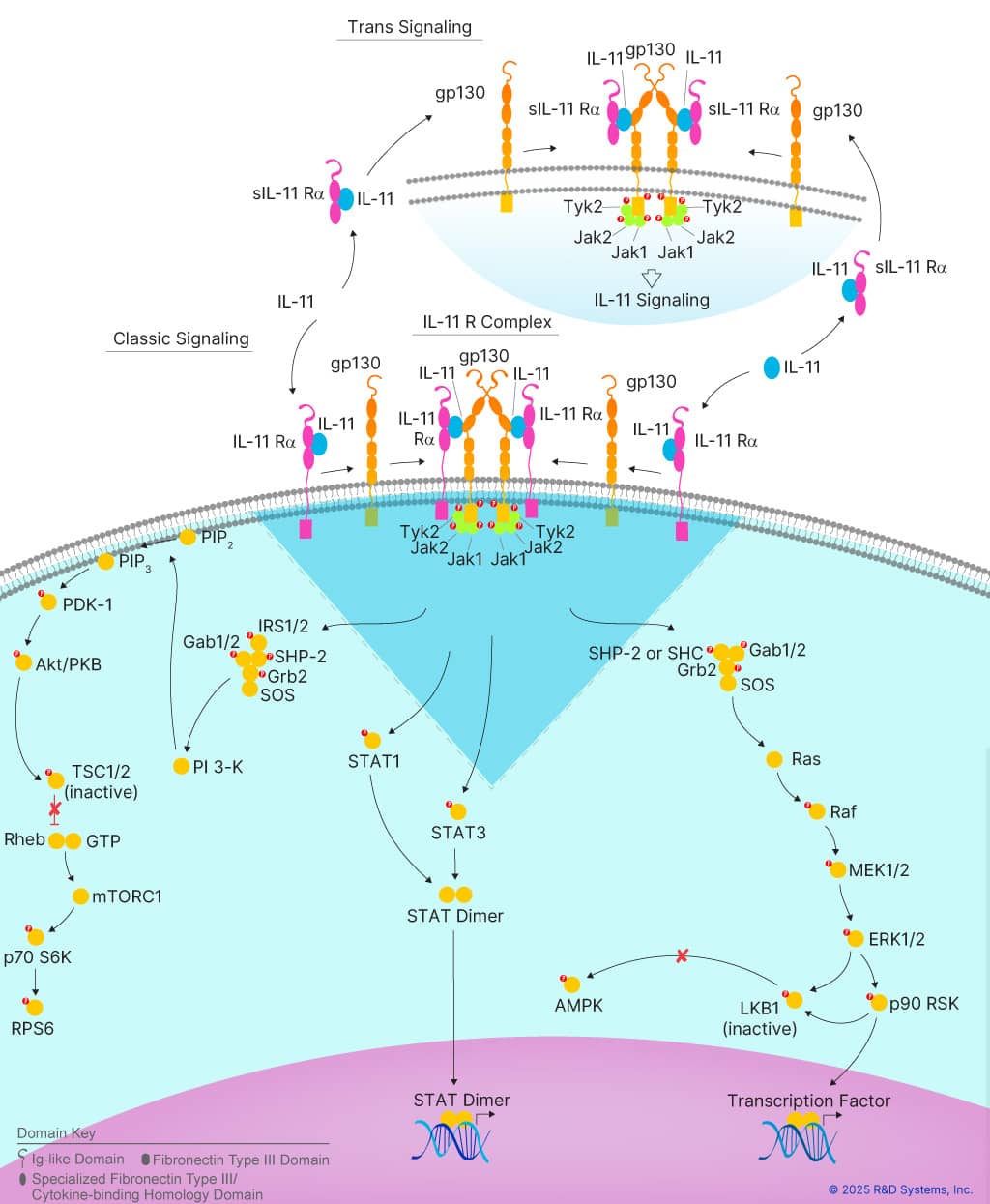
Figure 2. IL-11 Signaling Pathways. IL-11 signals through a receptor complex consisting of IL-11 Rα and gp130 to activate multiple intracellular signaling pathways, including the Jak-STAT, PI3K-Akt-mTORC1, and Ras-MEK-ERK pathways, which promote a pro-inflammatory state. Learn more about IL-11 Signaling Pathways here.
IL-11 Promotes ERK-mTORC1-Dependent Senescence
The recent study by Widjaja et al. showed that IL-11 expression is progressively upregulated with age in the liver, visceral gonadal white adipose tissue, and skeletal muscle of mice.5 This increase was accompanied by a gradual increase in the phosphorylation of mTOR, p70S6K, ERK, p90RSK, and LKB1, and a decrease in the LKB1-dependent phosphorylation of the metabolic regulator, AMPK.5, 8 To determine whether phosphorylation of these kinases was directly dependent on IL-11 signaling, phosphorylation was compared in young and old, wild-type and IL-11ra1-/- mice. While the authors observed an increase in the phosphorylation of ERK, p90RSK, mTOR, p70S6K, and LKB1, along with an increase in the levels of the canonical senescence markers, p16Inka4 and p21Cip1/Waf1 in old wild-type mice, the phosphorylation of these kinases and the levels of p16 and p21 were considerably lower in old IL-11ra1-/- mice, suggesting that IL-11 signaling promotes ERK-mTORC1-dependent cellular senescence.
This was tested directly by treating human fibroblasts and hepatocytes with IL-11, which again resulted in ERK and mTOR activation, increased levels of p16 and p21, and reduced expression of the cell cycle regulators, PCNA and cyclin D1. Significantly, these effects were prevented by the MEK inhibitor, U0126 and the mTORC1 inhibitor, rapamycin, supporting the conclusion that IL-11 activates ERK and mTORC1 to promote cellular senescence.
Disruption of IL-11 Signaling Inhibits Senescence and Delays Hallmarks of Aging
To further investigate the connection between IL-11 signaling and replicative senescence, the authors serially passaged human fibroblasts in the presence of an IL-11RA neutralizing antibody or an IgG control. They found that cells treated with an IL-11RA antibody had lower levels of passage-dependent ERK, mTOR, and STAT3 phosphorylation, coupled with lower expression of p16 and p21, and lower levels of inflammation-associated cytokines in cell culture supernatants, relative to fibroblasts passaged in the presence of the IgG control. IL-11RA antibody-treated late passage cells also had telomere lengths, mitochondrial DNA copy numbers, and oxygen consumption rates that were similar to the early passage cells, suggesting that disruption of IL-11 signaling could inhibit senescence and delay hallmarks of aging.
IL-11 Deletion Extends the Healthspan of Aging Mice
To explore the age-related effects of IL-11 signaling and support the results obtained with IL-11ra1-/- mice, young and old IL-11-/- mice were characterized. Like old IL-11ra1-/- mice, old IL-11-/- mice had lower levels of ERK, p90RSK, mTOR, p70S6K, and LKB1 phosphorylation than old wild-type mice, along with decreased levels of p16 and p21. Additionally, old IL-11-/- mice had longer telomeres and increased mitochondrial DNA content, coupled with reduced pro-inflammatory gene expression and lower serum IL-6 levels compared to old wild-type mice.
Regardless of gender, old IL-11-/- mice were found to have lower body weights and fat mass, increased lean mass, better frailty scores, and moderately higher body temperatures than old wild-type mice. Serum cholesterol and serum triglyceride levels that increased with age in wild-type mice, were reduced in old IL-11-/- mice. In addition, the response to glucose and insulin was similar in both old IL-11-/- mice and young wild-type mice, while old wild-type mice showed impaired glucose and insulin tolerance. Notably, skeletal muscle strength was higher in young and old IL-11-/- mice than in age-matched controls, and both male and female
IL-11-/- mice had reduced visceral white adipose tissue and inguinal subcutaneous white adipose tissue mass indices. Based on these results, the authors concluded that disruption of IL-11 signaling inhibits cellular senescence, protects against age-related metabolic decline, frailty, and obesity, and reduces age-associated inflammation.
Anti-IL-11 Therapy: The Key to a Longer, Healthier Life?
The authors went on to show that many of the beneficial effects associated with deletion of IL-11 in aging mice could be reproduced by treating mice from 75 to 100 weeks of age every three weeks with a neutralizing IL-11 antibody. Old mice treated with anti-IL-11 experienced a reduction in pro-inflammatory gene expression, a reduction in metabolic dysfunction, improved muscle strength, and a restoration of white adipose tissue beiging compared with those treated with an IgG control. Old IL-11-/- mice and old anti-IL-11-treated mice developed fewer macroscopic tumors than old wild-type mice, supporting the observation that inhibition of IL-11 signaling could improve the healthspan of aging mice.
Significantly, deletion of IL-11 or anti-IL-11 therapy was also found to dramatically extend the median lifespan of both female and male mice. While wild-type control mice had a median lifespan of 120.9 weeks, IL-11-/- mice had a median lifespan of 151 weeks, with a significant increase observed in both gender groups. This effect could be reproduced by treating 75-week old wild-type mice with monthly injections of an IL-11 neutralizing antibody until death. Anti-IL-11 treatment increased the lifespan of female mice from 117 weeks to 146 weeks, and it extended the lifespan of male mice from 130 weeks to 160 weeks.
The results of this study are extremely exciting as they show that anti-IL-11 therapy in older mice inhibits cellular senescence, improves healthspan, and at the same time, increases lifespan. While additional research is necessary to determine whether these findings are applicable to humans and other mammals, this study provides hope that anti-IL-11 therapy may have the potential to extend healthy aging and increase life expectancy in the future.
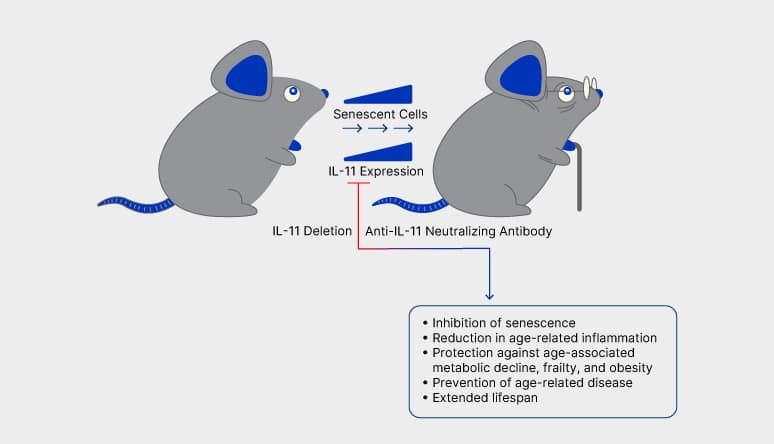
Figure 3. Deletion of IL-11 or Anti-IL-11 Therapy has Multiple Beneficial Effects on the Health and Longevity of Aging Mice. Widjaja et al. demonstrated that IL-11 expression is progressively upregulated in the tissues of aging mice and this increase is linked to an increase in cellular senescence and hallmarks of aging. Disruption of IL-11 signaling by either IL-11 deletion or anti-IL-11 therapy in old mice inhibited cellular senescence, reduced age-associated inflammation, protected against age-related metabolic decline, frailty, and obesity, and extended the lifespan of the mice.
References
- Li, X. et al. (2023) Signal Transduct Target Ther. 8:239.
- Karin, O. et al. (2019) Nat. Commun. 10:5495.
- von Kobbe, C. et al. (2019) Aging 11:12844.
- Baker, D.J. et al. (2016) Nature 530:184. *
- Widjaja, A. et al. (2024) Nature 362:157. *
- Ng, B. et al. (2020) Exp. Mol. Med. 52:1871.
- Pinti, M. et al. (2023) Int. J. Mol. Sci. 24:2719. *
- Widjaja, A. et al. (2022) iScience 25:104806. *
* Indicates publications that cite the use of products from R&D Systems or Novus Biologicals.