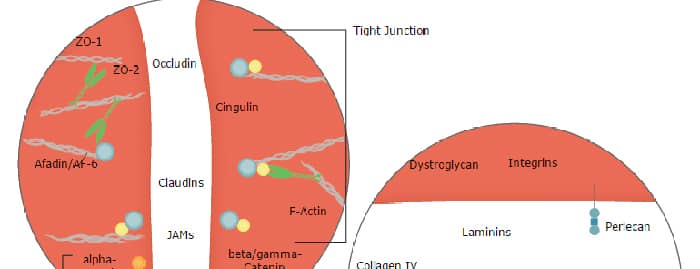
Dystroglycan, also called DAG‑1 (dystrophin‑associated glycoprotein 1) or DG, is a heterodimeric adhesion molecule that links the extracellular matrix (ECM) to the cell cytoskeleton (1‑4). Human preproDAG‑1 is an 895 amino acid (aa) type I transmembrane protein that contains a 27 aa signal sequence and an 868 aa proform. Autocatalysis of the proform produces two fragments that remain noncovalently‑linked. The first fragment (or alpha ‑chain) is 626 aa in length (aa 28‑653) and contains a mucin‑like region, while the second fragment (or beta ‑chain) is a 42‑44 kDa, 242 aa N‑glycosylated protein with an extracellular (aa 654‑749), transmembrane, and cytoplasmic domain (5). Over aa 28‑749, human DAG‑1 shares 93% aa sequence identity with mouse DAG‑1. It is widely expressed but differentially O‑glycosylated on skeletal muscle and epithelia (which contain a 160 kDa alpha ‑chain) as compared to cardiac muscle, smooth muscle, fibroblasts, keratinocytes, lymphocytes, and hematopoietic stem cells (which contain a 100 ‑ 140 kDa alpha ‑chain) (1‑3, 6‑9). DAG‑1 binding of ECM molecules is influenced by its alpha ‑chain O‑glycosylation (2, 6‑10). In addition to skeletal muscle and neuromuscular junctions in which DAG‑1 binds several ECM molecules, DAG‑1 is important for neuronal migration (through neurexin interactions), keratinocyte attachment to the ECM (through laminin), and adhesion at the immunological synapse and in the hematopoietic stem cell niche (through agrin) (3, 6‑11). In muscle, the beta ‑chain cytoplasmic domain connects with the cytoskeleton via formation of the dystrophin‑glycoprotein complex with isoforms of dystrophin, sarcoglycan, syntrophin, and sarcospan (3). This complex is critical for skeletal muscle viability and regeneration (3, 4, 10, 11). MMP9 cleavage of the 44 kDa beta ‑chain creates a 30 kDa transmembrane form that causes dissociation of the heterodimer and a down‑regulation of ECM interactions (6, 12). Dystroglycanopathies, a group of congenital muscular dystrophies affecting the brain, eye and skeletal muscle, are caused by either abnormalities in glycosyltransferases, or their accessory proteins, or rare DAG‑1 polymorphisms. All result in DAG‑1 hypoglycosylation, especially of O‑mannosyl forms, and affect DAG‑1 binding to ECM proteins (2, 3, 10, 13, 14).
Recombinant Human Dystroglycan Protein, CF
R&D Systems | Catalog # 6868-DG

Loading...
Key Product Details
- R&D Systems NS0-derived Recombinant Human Dystroglycan Protein (6868-DG)
- Quality control testing to verify active proteins with lot specific assays by in-house scientists
- All R&D Systems proteins are covered with a 100% guarantee
Source
NS0
Accession Number
Structure / Form
Noncovalently-linked heterodimer
Applications
Bioactivity
Loading...
Product Specifications
Source
Mouse myeloma cell line, NS0-derived human Dystroglycan protein
Met1-Val749, with a C-terminal 6-His tag
Met1-Val749, with a C-terminal 6-His tag
Purity
>95%, by SDS-PAGE visualized with Silver Staining and quantitative densitometry by Coomassie® Blue Staining.
Endotoxin Level
<0.10 EU per 1 μg of the protein by the LAL method.
N-terminal Sequence Analysis
Gln313 (blocked) of alpha chain and Ser654 of beta chain
Predicted Molecular Mass
67.8 kDa ( alpha subunit) & 11.3 kDa ( beta subunit)
SDS-PAGE
65-90 kDa & 20 kDa, reducing conditions
Activity
Measured by the ability of the immobilized protein to enhance the adhesion of H4 human neuroglioma cells.
The ED50 for this effect is 1.5-6.0 μg/mL.
Optimal dilutions should be determined by each laboratory for each application.
Formulation, Preparation, and Storage
6868-DG
| Formulation | Lyophilized from a 0.2 μm filtered solution in PBS. |
| Reconstitution | Reconstitute at 500 μg/mL in PBS.
Loading...
|
| Shipping | The product is shipped at ambient temperature. Upon receipt, store it immediately at the temperature recommended below. |
| Stability & Storage | Use a manual defrost freezer and avoid repeated freeze-thaw cycles.
|
Calculators
Background: Dystroglycan
References
- Ibraghimov-Bedkrovnaya, O. et al. (1993) Hum. Mol. Genet. 2:1651.
- Godfrey, C. et al. (2011) Curr. Opin. Genet. Dev. 21:278.
- Barresi, R. and Campbell, K.P. (2006) J. Cell Sci. 119:199.
- Durbeej, M. and K.P. Campbell (1999) J. Biol. Chem. 274:26609.
- Akhavan, A. et al. (2008) FASEB J. 22:612.
- Herzog, C. et al. (2004) J. Invest. Dermatol. 122:1372.
- Leonoudakis, D. et al. (2010) J. Cell Sci. 123:3683.
- Zhang, J. et al. (2006) FASEB J. 20:50.
- Mazzon, C. et al. (2011) Blood 118:2733.
- Michele, D.E. et al. (2002) Nature 418:417.
- Cohn, R.D. et al. (2002) Cell 110:639.
- Bozzi, M. et al. (2009) IUBMB Life 61:1143.
- Yoshida-Moriguchi, T. et al. (2010) Science 327:88.
- Hara, Y. et al. (2011) N. Eng. J. Med. 364:939.
Long Name
Dystrophin-associated Glycoprotein 1
Alternate Names
AGRNR, Dag-1, DAG1
Gene Symbol
DAG1
UniProt
Additional Dystroglycan Products
Product Documents for Recombinant Human Dystroglycan Protein, CF
Certificate of Analysis
To download a Certificate of Analysis, please enter a lot or batch number in the search box below.
Note: Certificate of Analysis not available for kit components.
Product Specific Notices for Recombinant Human Dystroglycan Protein, CF
For research use only
Related Research Areas
Citations for Recombinant Human Dystroglycan Protein, CF
 Powered by Bioz
Powered by Bioz
Customer Reviews for Recombinant Human Dystroglycan Protein, CF
There are currently no reviews for this product. Be the first to review Recombinant Human Dystroglycan Protein, CF and earn rewards!
Have you used Recombinant Human Dystroglycan Protein, CF?
Submit a review and receive an Amazon gift card!
$25/€18/£15/$25CAN/¥2500 Yen for a review with an image
$10/€7/£6/$10CAN/¥1110 Yen for a review without an image
Submit a review
Loading...
Associated Pathways