DNA Damage Response
Contents
- Introduction
- Cell Cycle Checkpoints
- G1 Checkpoint
- S-phase Checkpoint
- G2 Checkpoint
- DNA Repair Pathways
- References
Introduction
The maintenance of genome integrity and fidelity is essential for the proper function and survival of all organisms. This task is particularly daunting due to constant assault on the DNA by genotoxic agents (both endogenous and exogenous), nucleotide misincorporation during DNA replication, and the intrinsic biochemical instability of the DNA itself.1
Failure to repair DNA lesions may result in blockages of transcription and replication, mutagenesis, and/or cellular cytotoxicity.2 In humans, DNA damage has been shown to be involved in a variety of genetically inherited disorders, in aging,3 and in carcinogenesis.4, 5
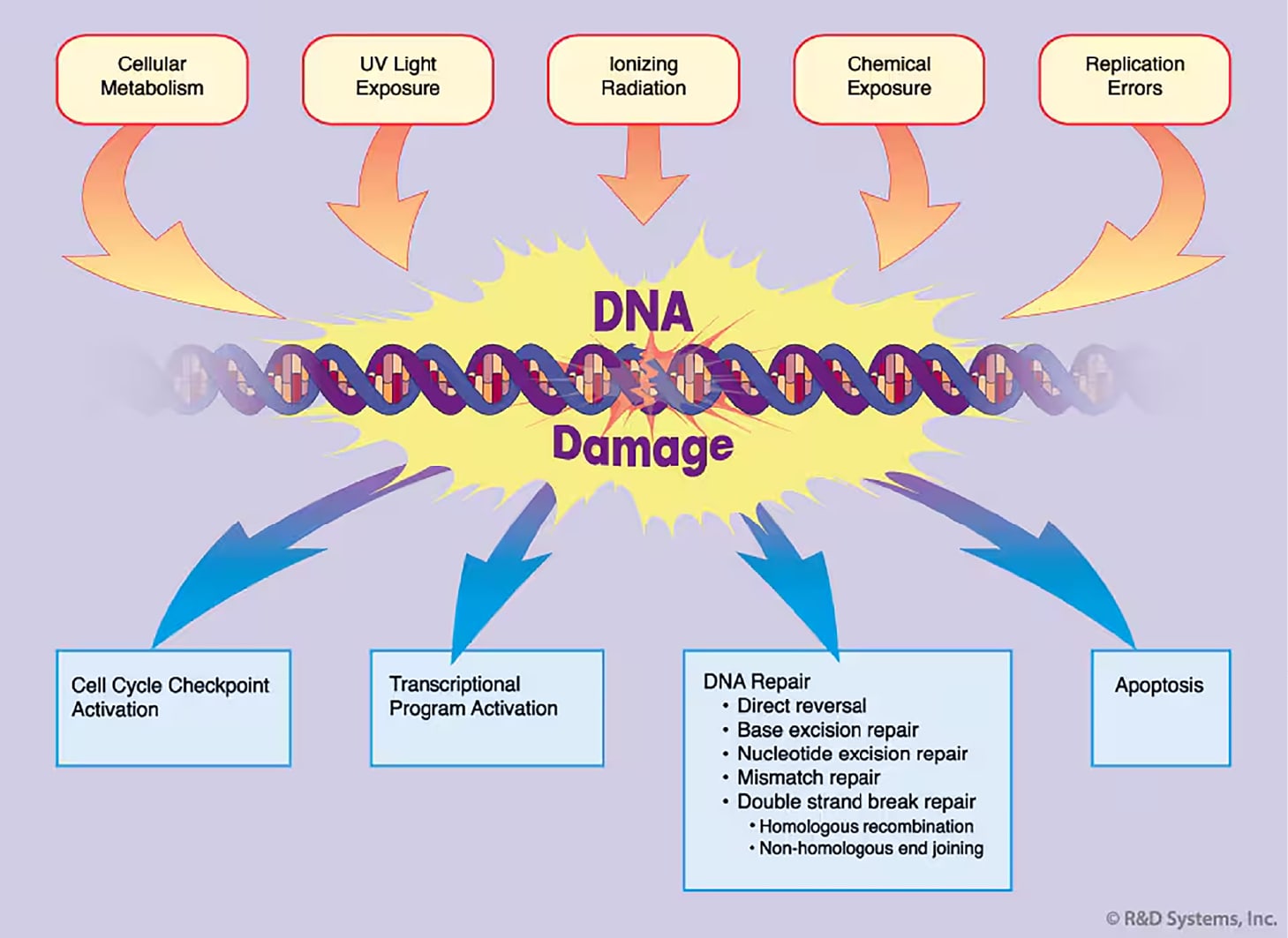
Figure 1. DNA Damage Response. DNA damage is caused by a variety of sources. The cellular response to damage may involve activation of a cell cycle checkpoint, commencement of transcriptional programs, execution of DNA repair, or when the damage is severe, initiation of apoptosis.
All eukaryotic cells have evolved a multifaceted response to counteract the potentially deleterious effects of DNA damage (Figure1).2 Upon sensing DNA damage or stalls in replication, cell cycle checkpoints are activated to arrest cell cycle progression to allow time for repair before the damage is passed on to daughter cells. In addition to checkpoint activation, the DNA damage response leads to induction of transcriptional programs, enhancement of DNA repair pathways, and when the level of damage is severe, to initiation of apoptosis.6 All of these processes are carefully coordinated so that the genetic material is faithfully maintained, duplicated, and segregated within the cell.
Cell Cycle Checkpoints
Cell cycle checkpoints are regulatory pathways that govern the order and timing of cell cycle transitions to ensure completion of one cellular event prior to commencement of another. The key regulators of the checkpoint pathways in the mammalian DNA damage response are the ATM (ataxia telangiectasia, mutated) and ATR (ATM and Rad3-related) protein kinases. Both of these proteins belong to a structurally unique family of serine-threonine kinases characterized by a C-terminal catalytic motif containing a phosphatidylinositol 3-kinase domain.7, 8 Although ATM and ATR appear to phosphorylate many of the same cellular substrates,9 they generally respond to distinct types of DNA damage. ATM is the primary mediator of the response to DNA double strand breaks (DSBs) that can arise by exposure to ionizing radiation (IR). ATR, on the other hand, plays only a back-up role in the DSB response, but directs the principle response to UV damage and stalls in DNA replication.
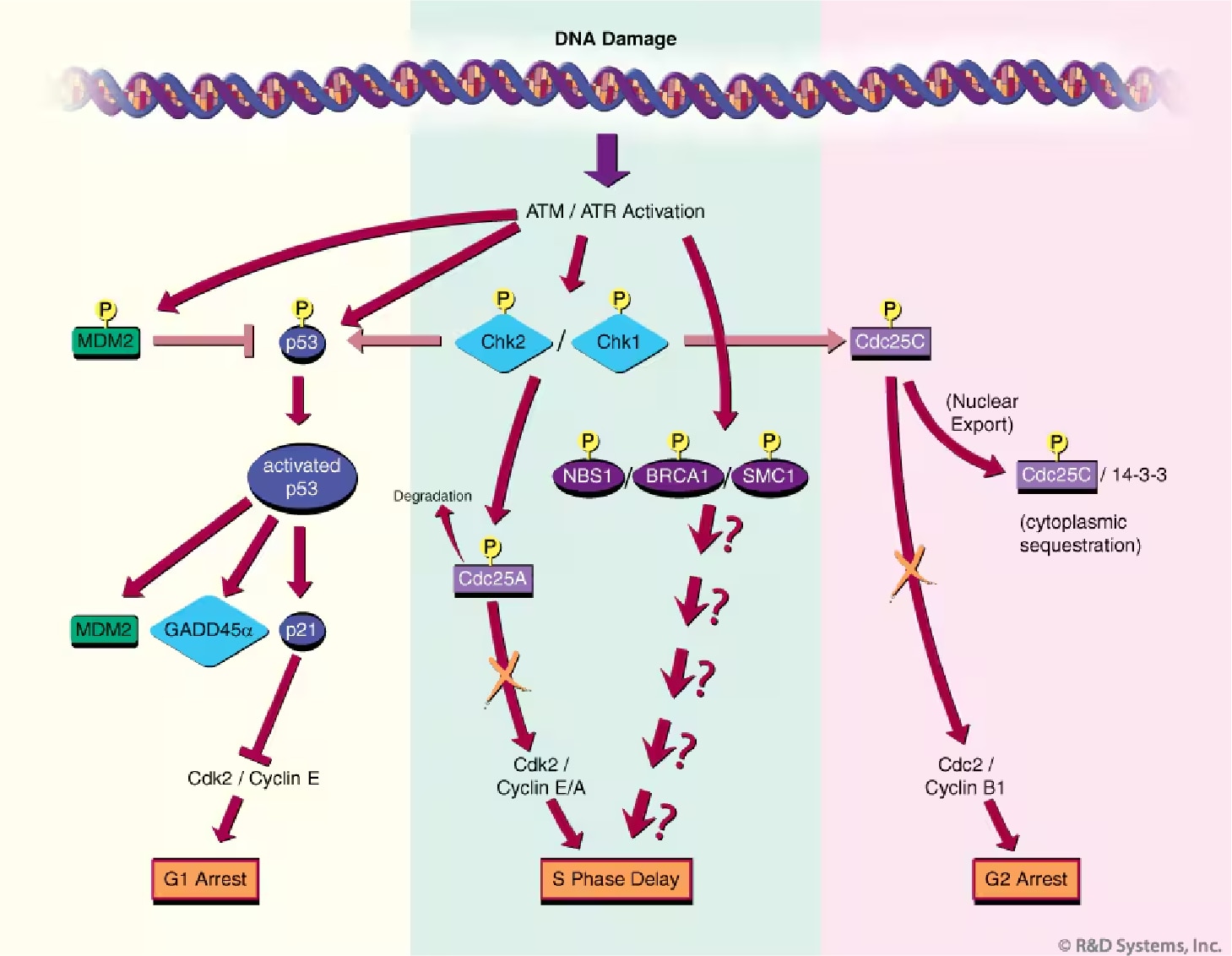
Figure 2. Mammalian Cell Cycle Checkpoint Pathways. In response to DNA damage, ATM and/or ATR trigger the activation of a checkpoint that leads to cell cycle arrest or delay. Checkpoint pathways are characterized by cascades of protein phosphorylation events (indicated with a "P") that alter the activity, stability, or localization of the modified proteins. A general overview of the G1, S, and G2 cell cycle checkpoint pathways is indicated (left, center, and right panels, respectively). See main text for additional details.
G1 Checkpoint
The G1 cell cycle checkpoint prevents damaged DNA from being replicated and is the best understood checkpoint in mammalian cells (Figure 2).10 Central to this checkpoint is the accumulation and activation of the p53 protein; two properties carefully controlled by the ATM and ATR kinases. In normally growing cells, p53 levels are low due to interaction with MDM2, which targets p53 for nuclear export and proteosome-mediated degradation in the cytoplasm.11 Following IR damage, ATM activates downstream kinase Chk2 (by phosphorylation at position T68),12 which in turn phosphorylates residue S20 of p53. The S20 phosphorylation of p53 blocks p53/MDM2 interaction, resulting in p53 accumulation. ATM exerts a second control measure on p53 stability by directly phosphorylating the p53 negative regulator, MDM2, on S395.13 This modification allows MDM2/p53 interaction, but prevents p53 nuclear export to the cytoplasm where degradation would normally occur. The role of ATR in p53 S20 phosphorylation (and subsequent stabilization) is less well established, but implied through in vitro evidence demonstrating S20 phosphorylation by the ATR-dependent kinase, Chk1.14
While phosphorylation of S20 is important to p53 stability, it is the phosphorylation of S15 that appears crucial in enhancing p53 transcriptional transactivation activity.15 The S15 residue of p53 can be phosphorylated directly by ATM or ATR in response to IR (ATM and ATR), UV irradiation (ATR) and stalls of DNA replication forks (ATR). Activated p53 then up-regulates a number of target genes, several of which are also involved in the DNA damage response (MDM2, GADD45a, and p21/Cip). The accumulation of p21, a cyclin-dependent kinase inhibitor, suppresses Cyclin E/Cdk2 kinase activity thereby resulting in G1 arrest (see reference 10 and references therein).
S-phase Checkpoint
The S-phase checkpoint monitors cell cycle progression and decreases the rate of DNA synthesis following DNA damage. Although this pathway is the least understood of the mammalian checkpoints, recent studies on IR-induced S-phase checkpoint activation are beginning to provide important insight. Cells from cancer-prone individuals affected with ataxia telangiectasia (AT) or Nijmegen breakage syndrome (NBS) fail to slow their rate of DNA replication following IR exposure; a phenomenon known as radio-resistant DNA synthesis (RDS). This finding implicates the associated gene products (ATM and NBS1, respectively) in the S-phase checkpoint pathway. Experimental evidence indicates that IR damage activates the S-phase checkpoint via at least 2 parallel branches, both of which are regulated by ATM.16, 17, 18 In the first branch, IR damage induces the phosphorylation of the Chk2 kinase (at T68) by ATM.19 Chk2, once activated, targets the Cdc25A phosphatase for ubiquitin-dependent degradation by phosphorylating it on S123.19 The resultant destabilization of Cdc25A prevents it from performing its normal function of removing inhibitory phosphorylations (T14 and Y15) from Cdk2. The Cdk2/Cyclin E and Cdk2/Cyclin A complexes remain inactive thus preventing completion of DNA synthesis.
The second branch of the IR-induced S-phase checkpoint pathway is independent of Cdc25A, but requires the activities of both ATM and NBS1.16, 17, 18 Upon IR damage, ATM phosphorylates a number of downstream substrates including NBS1 (at multiple sites including S343), the product of the breast cancer susceptibility gene 1 (BRCA1; at multiple sites including S1387), and SMC1 (structural maintenance of chromosome protein 1; at S957 and S966). Loss of any of these proteins or mutation of the indicated phosphorylation sites results in attenuated S-phase checkpoint activation.16, 17, 18, 20 Interestingly, the NBS1 and BRCA1 proteins are required for optimal phosphorylation of SMC1 upon IR,17, 18 perhaps suggesting that a larger protein complex must be formed before ATM can phosphorylate SMC1. Indeed, the ATM, NBS1, and BRCA1 proteins have all been shown to be part of a mega-dalton sized protein complex referred to as BASC (BRCA1- associated genome surveillance complex) that also includes numerous other DNA repair and replication factors.21 The precise roles of these proteins, and mechanistic details of how activation of this branch of the S-phase checkpoint leads to reduced DNA synthesis remain to be elucidated.
The involvement of ATR in the S-phase checkpoint also remains relatively obscure. ATR has been shown to initiate a slow IR- induced S-phase checkpoint response by phosphorylating its effector kinase, Chk1 (on S317 and S345), which in turn phosphorylates Cdc25A targeting it for degradation.22 In addition, SMC1 undergoes S957 and S966 phosphorylation upon UV irradiation or hydroxyurea treatment in an ATM-independent manner.18 The demonstrated responsiveness of ATR to UV damage and replication blocks make it the leading suspect in SMC1 phosphorylation under these conditions, however, experimental proof is lacking. Furthermore, an additional ATR-directed S-phase checkpoint pathway to deal with UV damage and replication errors has been reported.7
G2 Checkpoint
The G2 cell cycle checkpoint is an important control measure that allows suspension of the cell cycle prior to chromosome segregation. Entry into mitosis is controlled by the activity of the cyclin dependent kinase Cdc2.23 Maintenance of the inhibitory phosphorylations on Cdc2 (on T14 andY15) is essential for G2 checkpoint activation. ATM and ATR indirectly modulate the phosphorylation status of these sites in response to DNA damage. Unlike other checkpoints, the response to IR is mediated primarily by ATR24 with ATM playing a back-up role; the response to UV damage and replication blocks is controlled by ATR. It should be noted that the stage of the cell cycle when the DNA damage occurs may influence whether the response is mediated through ATR or ATM.7 In any case, upon DNA damage, the downstream kinases Chk1 and Chk2 (activated by ATR- and ATM-dependent phosphorylation, respectively) phosphorylate the dual specificity phosphatase Cdc25C on position S216.25 Phosphorylation of this residue creates a binding site for the 14-3-3 proteins. The 14-3-3/Cdc25C protein complexes are sequestered in the cytoplasm, thereby preventing Cdc25C from activating Cdc2 through removal of the T14 and Y15 inhibitory phosphorylations. This results in the maintenance of the Cdc2/Cyclin B1 complex in its inactive state and blockage of entry into mitosis.
DNA Repair Pathways
Direct Reversal The simplest of the human DNA repair pathways involves the direct reversal of the highly mutagenic alkylation lesion O6-methylguanine (O6-mG) by the product of the MGMT gene (O6-methylguanine DNA methyltransferase).26 The O6-mG adduct is generated in low levels by the reaction of cellular catabolites with the guanine residues in the DNA.27 Correction of the lesion occurs by direct transfer of the alkyl group on guanine to a cysteine residue in the active site of MGMT in a "suicide" reaction. The inactivated alkyl-MGMT protein is then degraded in an ATP-dependent ubiquitin proteolytic pathway.28 This energetically expensive repair mechanism for the correction of a relatively simple alkyl-adduct implies O6-mG is extremely detrimental to the cell. Accordingly, a number of chemotherapeutic agents that attack the O6 position of guanine have been developed and are in clinical use.29
BER Base excision repair (BER) is a multi-step process that corrects non-bulky damage to bases resulting from oxidation, methylation, deamination, or spontaneous loss of the DNA base itself.30 These alterations, although simple in nature, are highly mutagenic and therefore represent a significant threat to genome fidelity and stability.2
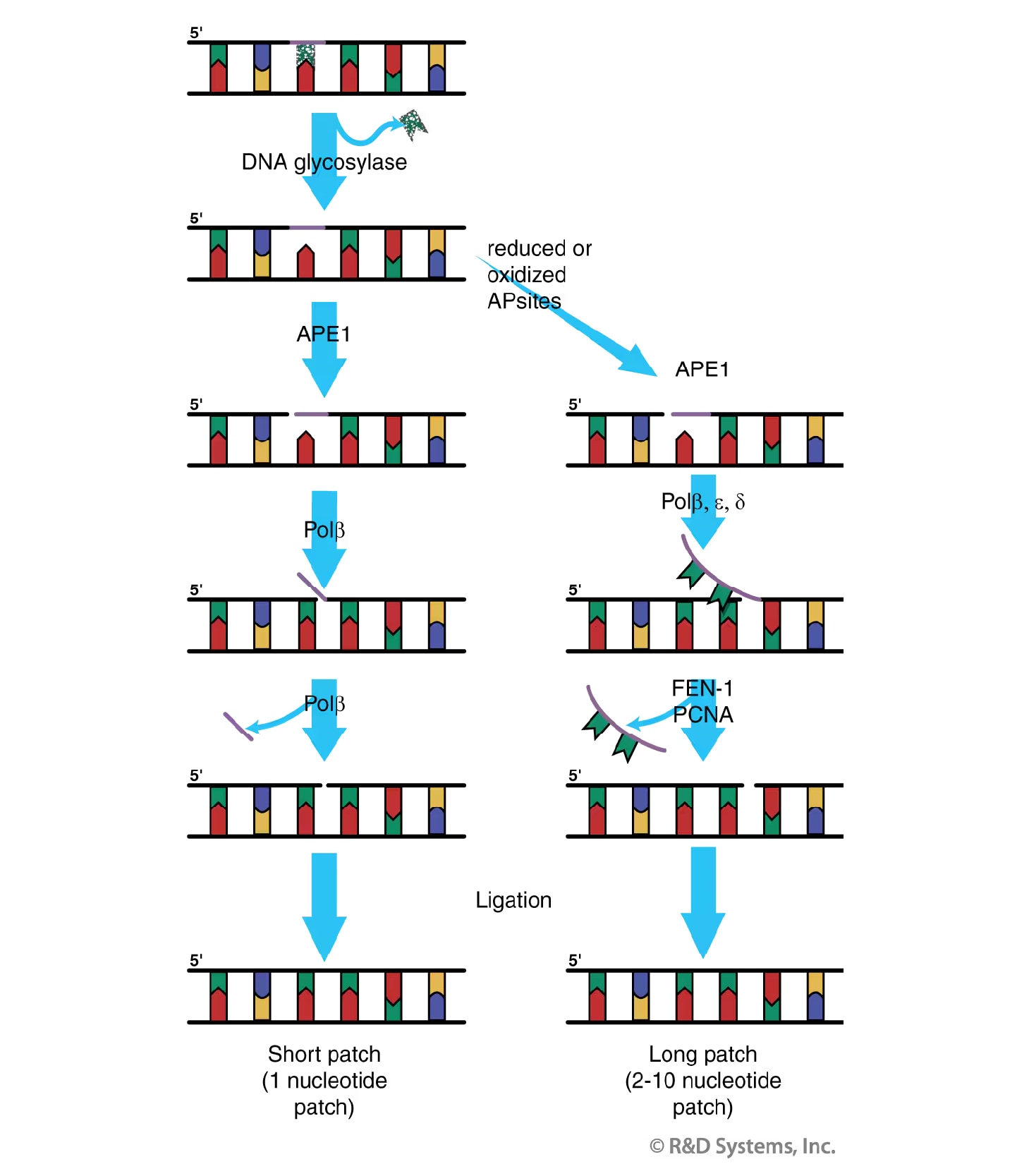
Figure 3. Base Excision Repair (BER). Shown is a general model of the short patch (left) and long patch (right) BER pathways. Short patch repair replaces the lesion with a single nucleotide; long patch repair replaces the lesion with approximately 2 to 10 nucleotides. See main text for additional details.
BER has two subpathways (Figure 3), both of which are initiated by the action of a DNA glycosylase that cleaves the N-glycosidic bond between the damaged base and the sugar phosphate backbone of the DNA. This cleavage generates an apyrimidinic/apurinic (AP) or abasic site in the DNA. Eight DNA glycosylases with partially overlapping base adduct specificity have been identified in humans.31 Alternatively, AP sites can also arise by the spontaneous hydrolysis of the N-glycosidic bond. In either case, the AP site is subsequently processed by AP Endonuclease 1 (APE1) which cleaves the phosphodiester backbone immediately 5' to the AP site, resulting in a 3' hydroxyl group and a transient 5' abasic deoxyribose phosphate (dRP). Removal of the dRP can be accomplished by the action of DNA polymerase beta (Pol b), which adds one nucleotide to the 3' end of the nick and removes the dRP moiety via its associated AP lyase activity.32 The strand nick is finally sealed by a DNA ligase, thus restoring the integrity of the DNA. Replacement of the damaged base with a single new nucleotide as described above is referred to as "short-patch" repair and represents approximately 80-90% of all BER.
The back-up pathway of BER, termed "long-patch" repair, is employed when a modified base resistant to the AP lyase activity of DNA Pol beta is present in the DNA.33 Long-patch repair results in the replacement of approximately 2-10 nucleotides including the damaged base. This subpathway requires many of the same factors involved in short-patch repair, including a DNA glycosylase, APE1 and DNA Pol beta. Unlike short-patch repair, however, long-patch repair is a PCNA-dependent pathway,34 where the DNA polymerase (beta d, zeta r epsilon) adds several nucleotides to the repair gap thus displacing the dRP as part of a "flap" oligonucleotide. The resulting oligonucleotide overhang is excised by the Flap endonuclease FEN-1 prior to sealing of the nick by a DNA ligase.
NER Nucleotide excision repair (NER) is perhaps the most flexible of the DNA repair pathways considering the diversity of DNA lesions it acts upon. The most significant of these lesions are pyrimidine dimers (cyclobutane pyrimidine dimers and 6-4 photoproducts) caused by the UV component of sunlight. Other NER substrates include bulky chemical adducts, DNA intrastrand crosslinks, and some forms of oxidative damage. The common features of lesions recognized by the NER pathway are that they cause both a helical distortion of the DNA duplex and a modification of the DNA chemistry.35
Considerable insight into the process of human NER has been gained through the study of two rare, autosomal recessive, but heterogeneous disorders - xeroderma pigmentosum (XP) and Cockayne Syndrome (CS). Individuals affected with either of these diseases display severe UV hypersensitivity. Both diseases are genetically heterogeneous; XP is caused by mutations in one of seven genes (XPA to XPG) and CS is caused by defects in one of two genes (CSA or CSB). The XP gene products are now known to perform various functions during damage recognition and DNA incision.36 The CS gene products, on the other hand, are required for NER-based repair of transcriptionally active genes.37
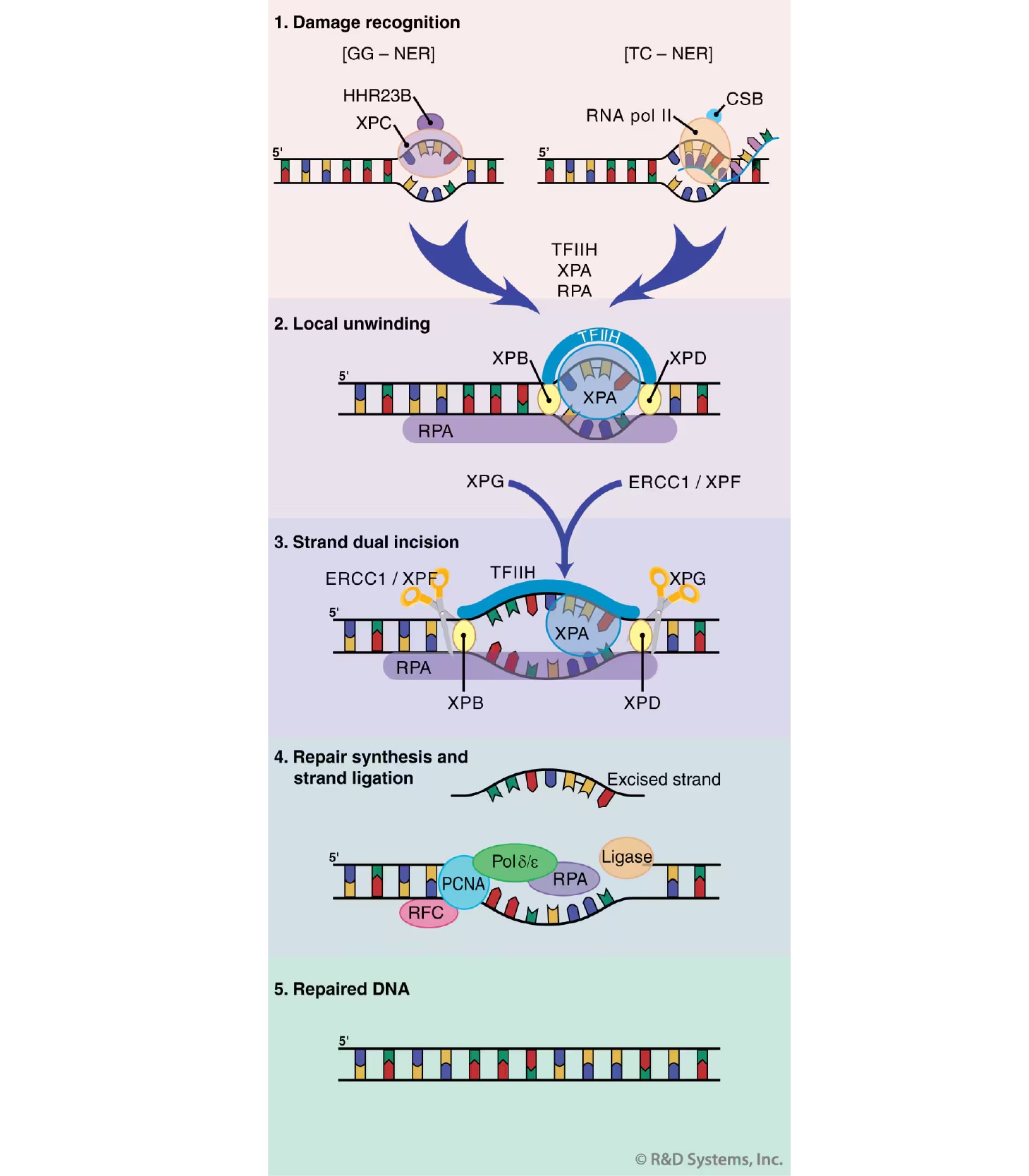
Figure 4. Nucleotide Excision Repair (NER). A simplified model of steps in NER is shown. DNA damage recognition (1) differs between global genomic and transcription coupled NER (GG- and TC-NER, respectively). The subsequent steps of these processes are shared and include local unwinding of the DNA (2), DNA strand dual incision (3), and DNA repair synthesis and strand ligation (4). See main text for additional details.
The NER process requires the action of more than 30 proteins in a stepwise manner that includes damage recognition, local opening of the DNA duplex around the lesion, dual incision of the damaged DNA strand, gap repair synthesis, and strand ligation (Figure 4).38 As alluded to above, there two distinct forms of NER: global genomic NER (GG-NER), which corrects damage in transcriptionally silent areas of the genome, and transcription coupled NER (TC-NER), which repairs lesions on the actively transcribed strand of the DNA. These two subpathways are fundamentally identical except in their mechanism of damage recognition. In GG-NER, the XPC/HHR23B protein complex is responsible for the initial detection of damaged DNA. Conversely, damage recognition during TC-NER does not require XPC, but rather is thought to occur when the transcription machinery is stalled at the site of injury. The stalled RNA polymerase complex must then be displaced in order to allow the NER proteins access to the damaged DNA. This displacement is aided by the action of the CSA and CSB proteins, as well as other TC-NER-specific factors. The subsequent steps of GG- and TC-NER proceed in an essentially identical manner. XPA and the heterotrimeric replication protein A (RPA) then bind at the site of injury and further aid in damage recognition. Next, the XPB and XPD helicases, components of the multi-subunit transcription factor TFIIH, unwind the DNA duplex in the immediate vicinity of the lesion. The endonucleases XPG and ERCC1/XPF then cleave one strand of the DNA at positions 3' and 5' to the damage, respectively, generating an approximately 30 base oligonucleotide containing the lesion. This oligonucleotide is displaced, making way for gap repair synthesis (performed by DNA Pol delta epsilon, as well as several replication accessory factors). Finally, the nick in the repaired strand is sealed by a DNA ligase, thus completing the NER process.
MMR The DNA mismatch repair (MMR) pathway plays an essential role in the correction of replication errors such as base-base mismatches and insertion/deletion loops (IDLs) that result from DNA polymerase misincorporation of nucleotides and template slippage, respectively. Mispairs generated by the spontaneous deamination of 5-methylcytosine and heteroduplexes formed following genetic recombination are also corrected via MMR. Defects in this pathway result in a so-called "mutator" cellular phenotype characterized by elevated frequencies in spontaneous mutations and increased microsatellite instability (MSI). Mutations in several human MMR genes cause a predisposition to hereditary nonpolyposis colorectal carcinoma (HNPCC), as well as a variety of sporadic tumors that display MSI.5
The overall process of MMR is similar to the other excision repair pathways (see long-patch BER, and NER), in that the DNA lesion (mismatch or IDL) is recognized, a patch containing the lesion is excised, and the strand is corrected by DNA repair synthesis and re-ligation (Figure 5).39 The mammalian MMR pathway is initiated by recognition of the mismatch or IDL by a heterodimer of the MSH2-MSH6 (known as MutS alpha beta) proteins. The predominant base-base mismatch and single-base IDL recognition activity in human cells is provided by MutS alpha;40 larger IDLs are usually recognized by MutS beta. There is, however, some partial overlap in substrate specificity by these two heterodimers indicating at least a minimal level of redundancy in substrate recognition by the MMR pathway.40 Following lesion identification, MutS alpha beta) binds ATP, undergoes a conformational shift, and translocates along the DNA away from the site of the lesion until additional MMR proteins are encountered.41, 42 Higher order protein complexes are formed, including those containing the heterodimeric proteins of MLH1-PMS2 (MutL alpha beta), MLH1-MLH3, and replication factors. Strand discrimination is thought to occur by MMR protein contact with the replication machinery.43 Support of this hypothesis is provided by the finding that MutL alpha can interact with replication accessory factor, PCNA.44 This interaction may provide a physical link between mismatch recognition and identification of the newly synthesized DNA strand at the replication fork. Excision and resynthesis of the nascent strand (containing the mismatch or IDL) is performed by a number of factors including PCNA, RPA, RFC, exonuclease I, DNA polymerases delta and epsilon, endonuclease FEN1, and additional factors.43
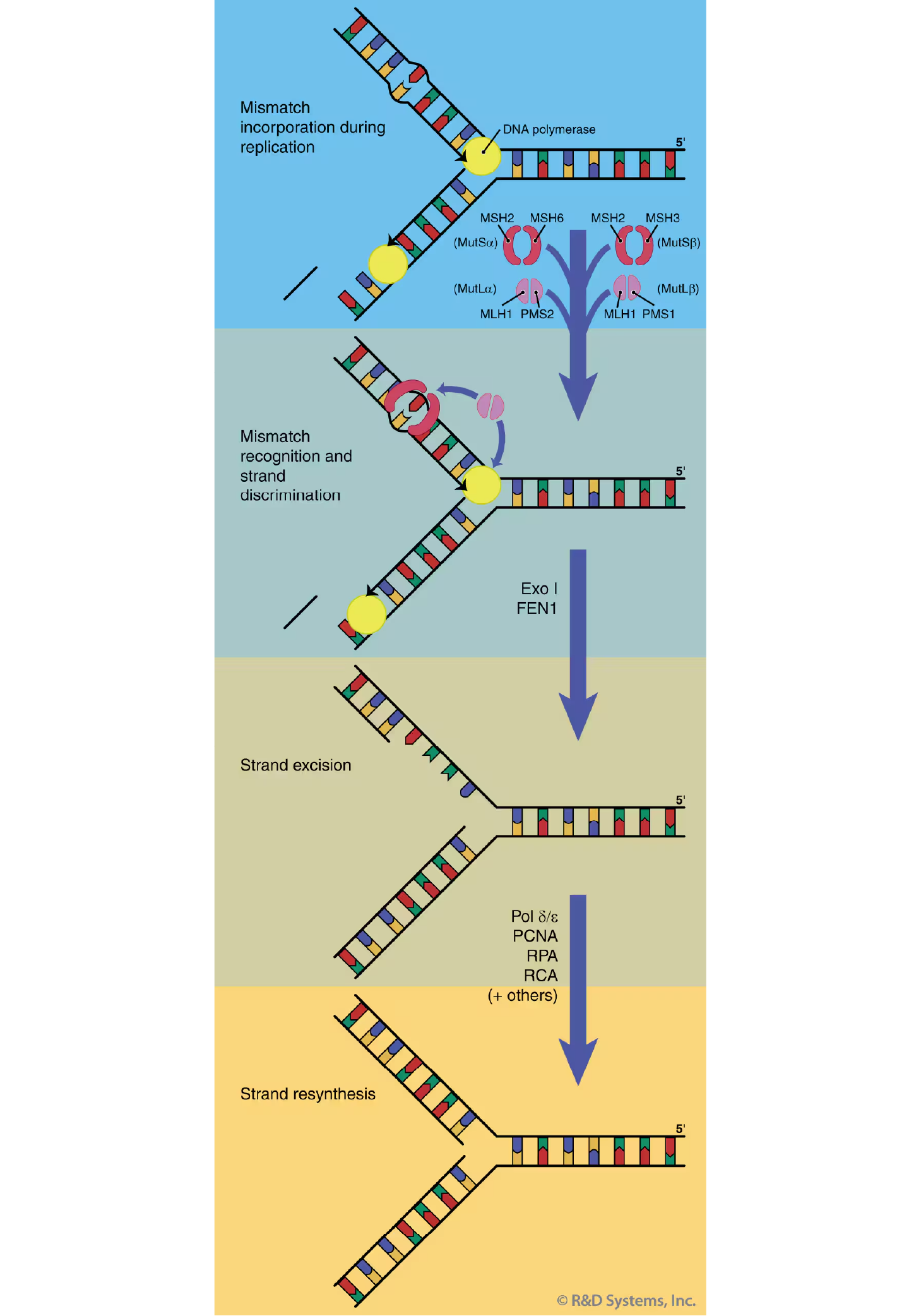
Figure 5. Mismatch Repair (MMR). A model of mammalian DNA MMR is shown. MMR is initiated by mismatch recognition and strand discrimination. Following excision of the newly synthesized strand containing the mismatched base or insertion/deletion loop (IDL), strand resynthesis is performed to restore the fidelity of the DNA.
DSB Repair Double-strand breaks (DSBs) are perhaps the most serious form of DNA damage because they pose problems for transcription, replication, and chromosome segregation. Damage of this type is caused by a variety of sources including exogenous agents such as ionizing radiation and certain genotoxic chemicals, endogenously generated reactive oxygen species, replication of single-stranded DNA breaks, and mechanical stress on the chromosomes. DSBs differ from most other types of DNA lesions in that they affect both strands of the DNA duplex and therefore prevent use of the complementary strand as a template for repair (see BER, NER, and MMR). Failure to repair these defects can result in chromosomal instabilities leading to dysregulated gene expression and carcinogenesis.4 To counteract the detrimental effects of these potent lesions, cells have evolved two distinct pathways of DSB repair,45 homologous recombination (HR) and non-homologous end joining (NHEJ) (Figure 6). The cellular decision as to which pathway to utilize for DSB repair is unclear, however, it appears to be largely influenced by stage within the cell cycle at the time of damage acquisition.46
HR-directed repair corrects DSB defects in an error-free manner using a mechanism that retrieves genetic information from a homologous, undamaged DNA molecule. The majority of HR-based repair takes place in late S- and G2-phases of the cell cycle when an undamaged sister chromatid is available for use as repair template. The RAD52 epistasis group of proteins, including RAD50, RAD51, RAD52, RAD54, and MRE11 mediate this process.47 The RAD52 protein itself is thought to be the initial sensor of the broken DNA ends. Processing of the damaged ends ensues resulting in the production of 3' single-stranded DNA (ssDNA) overhangs. The newly generated ssDNA ends are bound by RAD51 to form a nucleoprotein filament. Other proteins including RPA, RAD52, RAD54, BRCA1, BRCA2, and several additional RAD51-related proteins serve as accessory factors in filament assembly and subsequent RAD51 activities.45 The RAD51 nucleoprotein filament then searches the undamaged DNA on the sister chromatid for a homologous repair template. Once the homologous DNA has been identified, the damaged DNA strand invades the undamaged DNA duplex in a process referred to as DNA strand exchange. A DNA polymerase then extends the 3' end of the invading strand and subsequent ligation by DNA Ligase I yields a heteroduplexed DNA structure. This recombination intermediate is resolved and the precise, error-free correction of the DSB is complete.
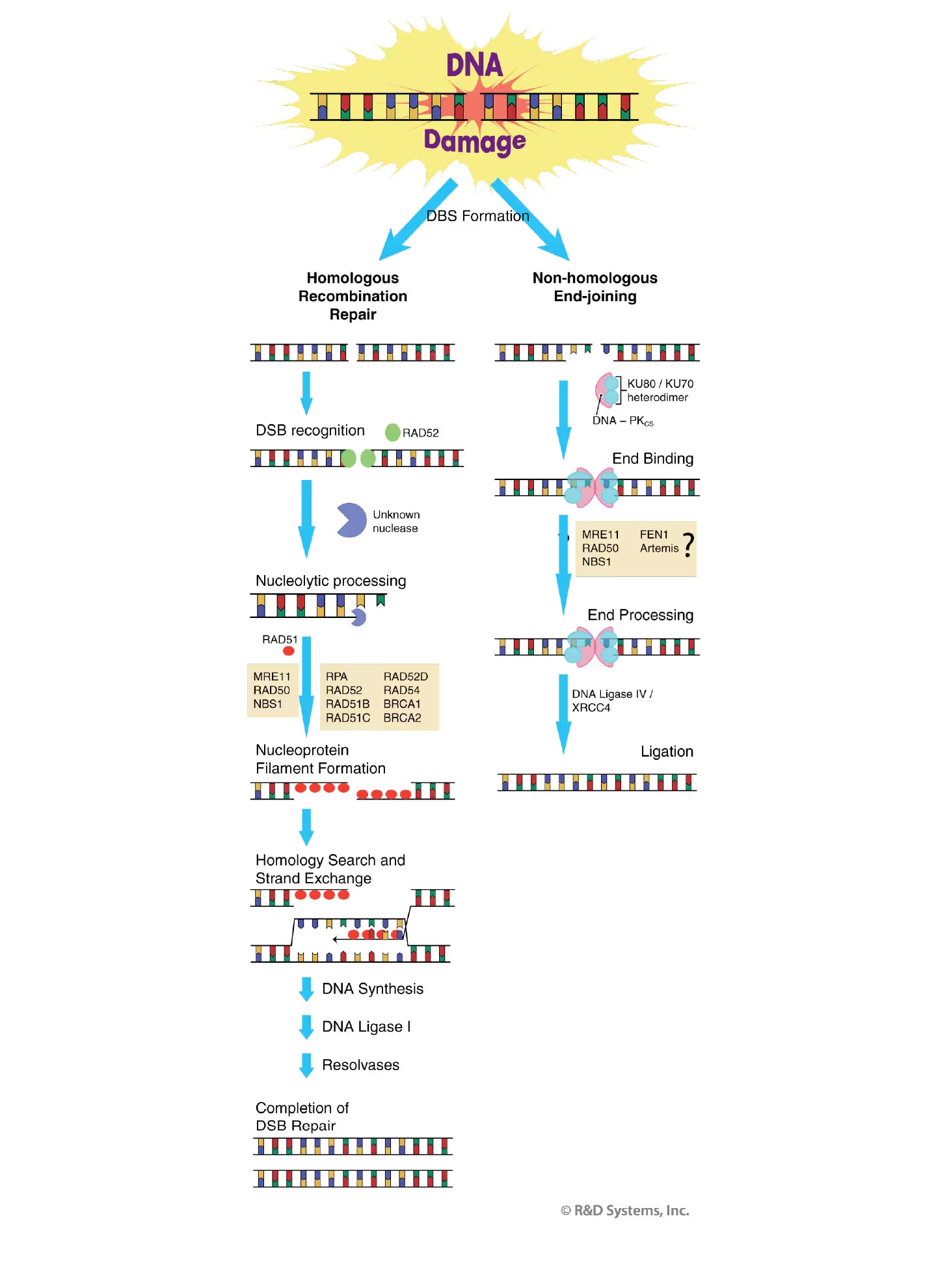
Figure 6. Double Strand Break (DSB) Repair. Shown is an overview of the main steps and factor requirements for DNA DSB repair by homologous recombination (left) and non-homologous end-joining (right). See main text for details.
In contrast to HR, NHEJ does not require a homologous template for DSB repair and usually results in the correction of the break in an error-prone manner. Essential to the NHEJ pathway is the activity of the Ku70/Ku80 heterodimeric protein.48 The Ku heterodimer initiates NHEJ by binding to the free DNA ends and recruiting other NHEJ factors such as DNA-dependent protein kinase (DNA-PK), XRCC4, and DNA Ligase IV to the site of injury.45 DNA-PK becomes activated upon DNA binding, and phosphorylates a number of substrates including p53, Ku, and the DNA Ligase IV cofactor XRCC4. Phosphorylation of these factors is believed to further facilitate the repair process. Because the ends of most DSBs generated by genotoxic agents are damaged and unable to be directly ligated, they often have to undergo limited processing by nucleases and/or polymerases before NHEJ can proceed. The nuclease(s) responsible for this processing remains to be determined, but strong candidates for this activity include the MRE11/Rad50/NBS1 complex,49, 50 FEN-1,51 and the Artemis protein.52 The final step in NHEJ repair involves ligation of the DNA ends by Ligase IV in a complex that also includes XRCC4 and Ku.
References
- Lindahl, T. (1993) Nature 362:709.
- Friedberg, E.C. et al. (1995) DNA Repair and Mutagenesis, ASM Press, Washington, DC.
- Finkel, T. & N.J. Holbrook (2000) Nature 408:239.
- Hoeijmakers, J.H.J. (2001) Nature 411:366.
- Peltomaki, P. (2001) Mutation Res. 488:77.
- Zhou, B.B. & S.J. Elledge (2000) Nature 408:433.
- Abraham, R.T. (2001) Genes Dev. 15:2177.
- Shiloh, Y. (2001) Curr. Opin. Genet. Dev. 11:71.
- Kim, S.T. et al. (1999) J. Biol. Chem. 274:37538.
- Bartek, J. & J. Lukas (2001) Curr. Opin. Cell. Biol. 13:738.
- Alarcon-Vargas, D. & Z. Ronai (2002) Carcinogenesis 23:541.
- Matsuoka, S. et al. (2000) Proc. Natl. Acad. Sci. USA 97:10389.
- Maya, R. et al. (2001) Genes Dev. 15:1067.
- Shieh, S.Y. et al. (2000) Genes Dev. 14:289.
- Dumaz, N. & D.W. Meek (1999) EMBO J. 18:7002.
- Falck, J. et al. (2002) Nature Genet. 30:290.
- Yardzi, P.T. et al. (2002) Genes Dev. 16:571.
- Kim, S.T. et al. (2002) Genes Dev. 16:560.
- Falck, J. et al. (2001) Nature 410:842.
- Xu, B. et al. (2002) Mol. Cell. Biol. 22:1049.
- Wang, Y. et al. (2000) Genes Dev. 14:927.
- Zhou X.Y. et al. (2002) Cancer Res. 62:1598.
- Nurse, P. (1990) Nature 344:503.
- Graves, P.R. et al. (2000) J. Biol. Chem. 275:5600.
- Peng, C.Y. et al. (1997) Science 277:1501.
- Margison, G.P. and M.F. Santibanez-Koref (2002) BioEssays 24:255.
- Sedgwick, B. (1997) Carcinogenesis 18:1561.
- Srivenugopal, K.S. et al. (1996) Biochemistry 35:1328.
- Gerson, S.L. (2002) J. Clin. Oncol. 20:2388.
- Memisoglu, A. & L. Samson (2000) Mutation Res. 451:39.
- Sharer, O.D. & J. Jiricny (2001) BioEssays 23:270.
- Matsumoto, Y. & K. Kim (1995) Science 269:699.
- Matsumoto, Y. et al. (1994) Mol. Cell. Biol. 14:6187.
- Frosina, G. et al. (1996) J. Biol. Chem. 271:9573.
- Hess, M.T. et al. (1997) Proc. Natl. Acad. Sci. USA 94:6664.
- Lindahl, T. & R.D. Wood (1999) Science 286:1897.
- Friedberg, E.C. (1996) BioEssays 18:731.
- Batty, D.P. & R.D. Wood (2000) Gene 241:193.
- Marti, T.M. et al. (2002) J. Cell. Physiol. 191:28.
- Genschel, J. et al. (1998) J. Biol. Chem. 273:19895.
- Gradia, S. et al. (1999) Mol. Cell 3:255.
- Blackwell, L.J. et al. (1998) J. Biol. Chem. 273:32055.
- Buermeyer, A.B. et al. (1999) Annu. Rev. Genet. 33:533.
- Umar, A. et al. (1996) Cell 87:65.
- Jackson, S.P. (2002) Carcinogenesis 23:687.
- Takata, M. et al. (1998) EMBO J. 17:5497.
- Sonoda, E. et al. (2001) Proc. Natl. Acad. Sci. USA 98:8388.
- Featherstone, C. & S.P. Jackson (1999) Mutat. Res. 434:3.
- Haber, J.E. (1998) Cell 95:583.
- Petrini, J.H.J. (2000) Curr. Opin. Cell Biol. 12:293.
- Wu, X. et al. (1999) Proc. Natl. Acad. Sci. USA 96:1303.
- Moshous, D. et al. (2001) Cell 105:177.
Related Information
DNA Damage, Mutation Detection and Enzyme Kits/Activity Assays
Genotoxic Stress Response/Cell Cycle
Immunoglobulin-Like Domain Transcription Factors