Apoptosis
Apoptosis, a form of programmed cell death, is a normal biological process that is required for proper organ development during embryogenesis and the removal of abnormal cells, such as cells that are damaged by exposure to pathogens or undergo oncogenic transformation. The switch between cell survival and apoptosis is tightly regulated and critical to the development and well-being of an organism. R&D Systems offers a range of products for studying apoptosis including proteins, antibodies, small molecules, kits, arrays, and more.
What is Apoptosis?
Apoptosis is a form of programmed cell death characterized by a coordinated and step-wise series of biochemical reactions that result in the ordered disassembly of a cell within an organism. It can be initiated via several mechanisms including trophic factor withdrawal, ligation of pro-apoptotic transmembrane receptors, or the activities of cytotoxic immune cells, which lead to the activation of intracellular proteases, endonucleases, and other enzymes that cleave key target substrates to dismantle the cell.
Significant cellular changes characteristic of apoptosis include:
- Cytoplasmic condensation and cell shrinkage
- Changes in the phospholipid content of the outer leaflet of the plasma membrane
- Disruption of the electrochemical gradient across the mitochondrial membrane
- Chromatin condensation and DNA degradation
- Membrane blebbing
- Formation of apoptotic bodies that can be efficiently cleared by phagocytes
Cytology of Apoptosis
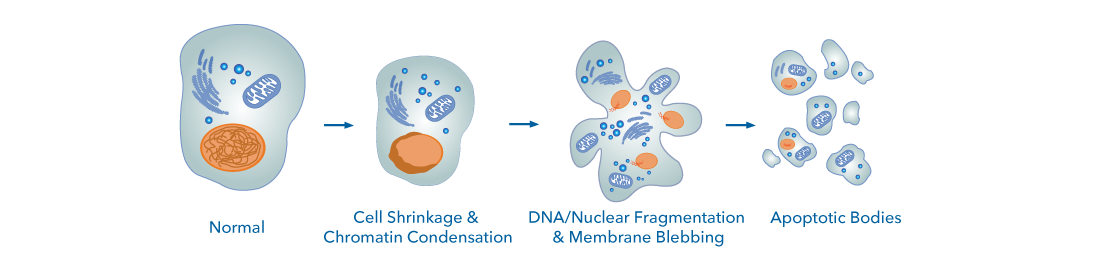
Figure 1. Morphological Changes Associated with Apoptosis. Induction of apoptosis evokes several significant phenotypic alterations in a cell, including cell shrinkage and condensation, DNA/nuclear fragmentation, membrane blebbing, and the formation of apoptotic bodies, which are cleared by phagocytes and neighboring cells.
Defects in the apoptotic pathway that prevent cell death may lead to developmental abnormalities or unregulated tissue growth, as occurs in cancer. In contrast, pathological increases in apoptotic activity are hallmarks of several disease states, including neurodegenerative disorders, insulin-dependent diabetes, myocardial infarction, atherosclerosis, and AIDS. Consequently, manipulation of the apoptotic process is essential to better understand the development of various diseases and to discover potential therapeutic targets.
Apoptosis Signaling
Apoptosis occurs via complex signaling cascades that trigger several significant changes and eventually lead to cell death. These molecular events can be used as potential biomarkers of apoptosis. Learn more about the cellular changes that occur during apoptosis and the small molecule apoptosis activators and inhibitors that are useful tools for manipulating and exploring apoptosis signaling.
Other Apoptosis Molecules
Induce Apoptosis with Ease
Apoptosis inducing compounds exert pro-apoptotic effects via a variety of mechanisms including DNA cross-linking, inhibition of anti-apoptotic proteins, and caspase activation.
Simultaneously Detect Apoptosis-related Proteins
Rapid, sensitive, all-in-one kit for detecting the relative expression of up to 35 apoptosis-related proteins in a single sample.
Simplify Apoptosis Detection
Choose from in situ detection of apoptosis in fixed frozen, paraffin-embedded, or plastic-embedded cells and tissues OR identify and quantify cells in suspension by flow cytometry.
Products for Inducing and Detecting Apoptosis
| Annexin V Kits | ApoStat Apoptosis Detection Kit | Apoptosis Antibody Arrays |
| Apoptosis Inducers and Inhibitors | Cell Proliferation Viability Apoptosis Assays | TUNEL Assays |
Apoptotic Events
Apoptosis is a dynamic process and determining the optimal time to measure the incidence of apoptosis or the activity of the molecules involved is critical for understanding the underlying mechanisms. Several factors can influence the timing and should be considered in the experimental design, including cell type, apoptotic stimulus, the specific apoptotic mechanism, and experimental conditions.

Several factors can affect how cells respond to an apoptosis-inducing agent and so it is important to understand the biological system under study. Primary cells may respond to death stimuli differently compared to immortalized cells, and cancer cell lines may respond differently than primary tumors or freshly explanted cells. For example, the human breast adenocarcinoma cell line, MCF-7, lacks Caspase-3, while primary neurons do not express the Bcl-2 family member Bak, depending almost exclusively on Bax for mitochondria-mediated apoptosis.