Molecular Balance in the Brain - R&D Systems
APP Cleavage & A-beta degradation
Neuroscience research suggests a delicate balance between physiological and pathological events in the brain. Healthy neurons predominantly process beta-amyloid precursor protein (APP) via non-amyloidogenic alpha-secretase-driven cleavage. The majority of beta-amyloid peptide (A-beta) generated is rapidly degraded by a range of enzymes expressed in neurons and supporting glia. In viable neurons, cytoskeletal proteins such as Tau, maintain an ordered intracellular scaffolding and support dendritic spines. Intraneuronal Ca2+ levels are tightly controlled and anti-apoptotic signaling preserves mitochondrial integrity. Under normal physiological conditions, astrocytes secrete trophic factors, such as NGF, BDNF, and GDNF, which have positive effects on neuronal viability. The release of IL-4, IL-10, and TGF-beta by macrophages also maintains the balance between pro- and anti-inflammatory cytokine signaling. Although soluble forms of A-beta have been shown to have a variety of effects on neuronal function, a physiological role for this peptide remains controversial. Low levels of A-beta are deposited as diffuse plaques in the brains of subjects without obvious cognitive dysfunction. Whether this accumulation of A-beta represents a part of “normal” aging or a very early sign of disease, has yet to be determined.
APP Metabolism
Under normal conditions, the majority of amyloid precursor protein (APP) cleavage is believed to be alpha-secretase-dependent and occurs within the amyloid-beta peptide (A-beta) sequence. However, changes that occur during aging and neurodegenerative diseases drive an increased proportion of amyloidogenic processing (BACE-1, beta secretase). R&D Systems offers a broad range of products designed for the study of APP metabolism, including recombinant APP-directed proteases, antibodies, ELISA-based activity assays, and more. We also offer products to study molecules which have been shown to modulate APP function and cleavage, such as Apolipoprotein E (ApoE), F-Spondin, G Protein-Coupled Receptor 3 (GPR-3), Lipoprotein Receptors (LRP), Netrin, Nogo, and Reelin.
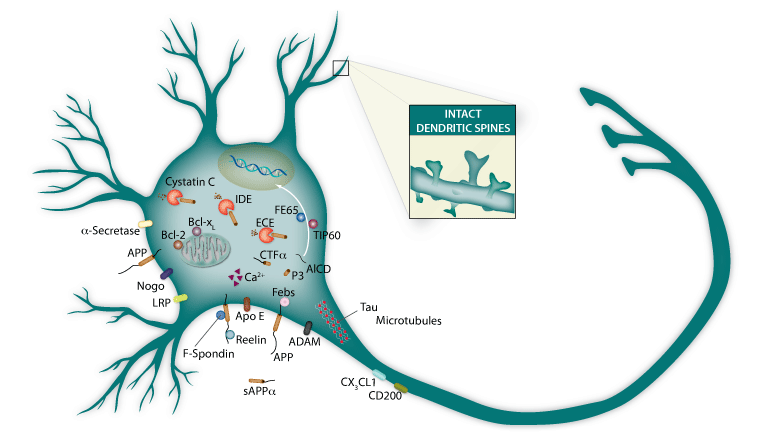
Figure 1. Non-amyloidogenic APP processing via alpha-secretase. Under normal conditions the vast majority of APP is cleaved by alpha-secretase to generate sAPP-alpha and C-terminal Fragment-alpha (CTF-alpha). CTF-alpha can be further cleaved by gamma-secretase to yield P3 and APP Intracellular Domain (AICD). The relatively small levels of A-beta produced are rapidly metabolized by A-beta degrading enzymes, such as IDE, ECE, and Cystatin C. View a complete list products related to APP Cleavage & Amyloid-beta Degradation.
A-beta Degradation
Age-dependent accumulation of A-beta in the brain is dependent on the rates of generation and degradation. A-beta peptides within the cell are subjected to degradation by Endothelin-Converting Enzyme (ECE), Insulin-Degrading Enzyme (IDE), and Cystatin-C. Breakdown of extracellular A-beta peptides is achieved through Neprilysin (NEP), Angiotensin Converting Enzyme (ACE), Matrix Metalloproteinases (MMP), and via Plasmin. Endocyotosis of A-beta molecules in macrophages and glia is facilitated through proteins such as alpha2-macroglobulin, Scavenger Receptor-A1 (SR-A1), Formyl Peptide Receptor-like Receptor (FPRL), Low-density Lipoprotein Receptor (LDL R), and LDL R-related protein-1 (LRP-1).

Figure 2. Degradation of extracellular A-beta by glial cells prevents amyloid plaque formation. In a healthy non-diseased brain the small amount of extracellular A-beta is degraded by microglia and astrocytes expressing ECE, NEP, and MMPs. A-beta peptides are internalized through LRP1, LDL R, and SR-A1. In addition, macrophages degrade A-beta molecules through alpha-2-macroglobulin uptake and MMP proteolysis. View a complete list products related to APP Cleavage & Amyloid-beta Degradation.
Neuronal Aging & A-beta Accumulation
Aging and disease exert molecular challenges on neuronal health. An increased proportion of APP is sequentially cleaved by BACE and beta-secretase, releasing higher levels of A-beta. Increased accumulation of A-beta molecules is thought to initiate a variety of pathological events which compromise neuronal viability and function. A-beta molecules interact with neurotransmitter receptors, disrupt mitochondrial function, impair Ca2+ homeostasis, and promote pro-apoptotic signaling. Increased kinase activation results in hyper-phosphorylation of tau, cytoskeletal compromise, collapse of dendritic spines, and formation of neurofibrillary tangles. Injured neurons release a variety of factors that induce inflammation and free radical generation, the effects of which are exacerbated by the release of pro-inflammatory cytokines. The accumulation of A-beta as mature plaques was originally considered to be the primary causative event during Alzheimer’s disease. However, recent studies focused on oligomerization of A-beta suggest that potent dysfunctional effects of this peptide may precede the more obvious structural changes.
Aberrant Cell Signaling & Apoptosis
Accumulating A-beta molecules exert a myriad of deleterious effects on the surrounding neurons and glia. A-beta peptides have been shown to induce neuronal dysfunction following interaction with cell surface receptors including NDMA R, TNF R, and the Receptor for Advanced Glycosylation End-products (RAGE). Neurotoxic effects of A-beta are known to include generation of reactive oxygen species (ROS), disruption of the cytoskeleton, induction of apoptotic cascades, and promotion of excitotoxicity. Increased expression of pro-apoptotic Bcl-2 family proteins induce pore formation in mitochondrial membranes and the release of pro-apoptotic Cytochrome c.
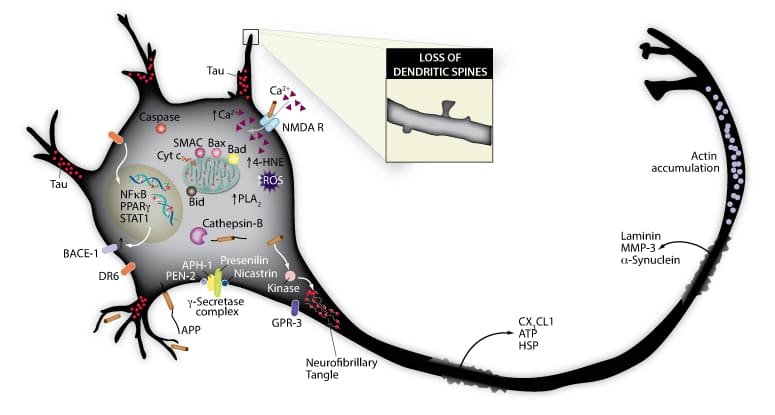
Figure 3. A-beta generation challenges neuronal viability and promotes apoptosis. Increased amyloidogenic processing of APP by BACE-1 and the gamma-secretase complex results in augmented levels of A-beta. In vulnerable neurons, A-beta peptides promote apoptosis, compromise mitochondrial integrity, and induce free radical generation, leading to the release of injury markers. A-beta is also thought to promote kinase activity, tau phosphorylation, the formation of neurofibrillary tangles, and the loss of dendritic spines. View a complete list products related to Apoptosis.
Cytokine-Induced Neuroinflammation
Cytokines play an important role in the brain under normal conditions and during the progression of disease. The release of anti-inflammatory cytokines, such as IL-4, IL-10, and TGF-beta enhance degradation of A-beta molecules through phagocytosis and receptor-mediated uptake. Paradoxically, chronic exposure to pro-inflammatory mediators may exacerbate the neurodegenerative environment. Enhanced release of pro-inflammatory cytokines further compromises vulnerable neurons, by increasing the release of injury markers and promoting prolonged release of IL-1beta, IL-6, and TNF-alpha.
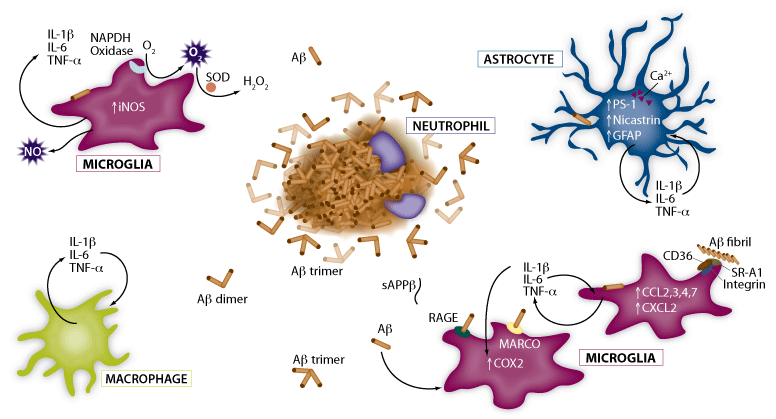
Figure 4. Accumulation of A-beta as mature plaques and associated inflammation. During Alzheimer’s disease (AD), A-beta molecules accumulate as mature A-beta plaques. In response to the deposition of A-beta and the release of chemoattractants from damaged neurons, activated astrocytes, microglia, and macrophages release pro-inflammatory cytokines such as IL-1beta, IL-6, and TNF-alpha. Although this initial response is an attempt to protect the brain, the prolonged state of chronic inflammation is believed to be a major detrimental factor during AD. View a complete list of products related to Neuroinflammation.
