Protocol for Culturing Embryonic Rat Spinal Motor Neurons
The spinal motor neuron culture is an indispensable model system for studying neuronal development, regeneration, and the mechanisms underlying motor neuron diseases such as amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA). Motor neuron survival in vitro requires a specific combination of multiple growth factors and supplemental reagents. This protocol describes the use of these growth factors and supplemental reagents for successful culture of motor neurons in vitro together with a simple step-by-step method for isolating and culturing these neurons.
Note: Aseptic techniques should be used in this protocol to ensure there is no bacterial, fungal, or mycoplasma contamination. The initial dissection and collection of the spinal cords can be completed outside of a laminar flow cell culture hood. However, preparation of cell culture plates and all steps following tissue harvest should be conducted within a hood. Likewise, all reagents and materials used should be sterile.
Supplies Required
Note: Cold Spring Harbor Protocols provides a recipe for the SATO supplement.1 The N-2 MAX Media Supplement (R&D Systems, Catalog # AR009) offers a serum-free alternative to the SATO supplement.
Note: Insulin has low solubility at a neutral pH. Dilute HCl acid (e.g. 1 N) can be added to the solution to help solubilize Insulin.
Reagents
- N-acetyl-L-cysteine solution (5 mg/mL in Neurobasal® medium)
- Bovine Serum Albumin (BSA, 4% in DPBS)
- Cultrex® Mouse Laminin I (R&D Systems, Catalog # 3400-010-02)
- Cultrex® Poly-D-Lysine (R&D Systems, Catalog # 3439-200-01)
- Deionized or distilled H2O, sterile (dH2O)
- Dimethyl sulfoxide (DMSO)
- DPBS, no Ca2+, no Mg2+ (ThermoFisher Scientific, Catalog # 14190250), or equivalent
- Fetal bovine serum (FBS)
- Forskolin solution (4.2 mg/mL in DMSO)
- L-glutamine-penicillin-streptomycin solution (100x), or equivalent
- Insulin solution (0.5 mg/mL in dH2O)
- Isobutylmethylxanthine solution (1 mg/mL in DMSO)
- Leibovitz’s L-15 Medium (ThermoFisher Scientific, Catalog # 14415064), or equivalent
- N21-MAX Media Supplement (50x, R&D Systems, Catalog # AR008)
- NaOH (0.1 N)
- Neurobasal® medium (ThermoFisher Scientific, Catalog # 21103049), or equivalent
- OptiPrep™ Density Gradient Medium (Sigma-Aldrich, Catalog # D1556), or equivalent
- Recombinant Human BDNF (R&D Systems, Catalog # 11166-BD)
- Recombinant Human CNTF (R&D Systems, Catalog # 257-NT)
- Recombinant Human GDNF (R&D Systems, Catalog # 212-GD)
- SATO supplement (100x), or equivalent
- Sodium pyruvate (100 mM)
- 3,3,5-triiodo-L-thyronine (T3) sodium salt solution (4 mg/mL)
- Trypan blue (0.4%)
- TrypZean™ solution (1x, Sigma-Aldrich, Catalog # T 3449), or equivalent
Optional Reagent
- Cytosine arabinoside (Sigma-Aldrich, Catalog # C1768)
Materials
- 0.22 µm sterile filter unit
- 15 mL conical centrifuge tubes, sterile
- 50 mL conical centrifuge tubes, sterile
- Cell culture plates (24-well), sterile
- E14–E15 timed pregnant rat
- Ice
- Parafilm®
- Pasteur pipette, glass, fire-polished, sterile
- Petri dishes, 60 × 15 mm
- Petri dishes, 100 × 20 mm
- Pipette tips
- Sylgard®-lined dissection petri dish, 93 × 22 mm (Living Systems Instrumentation, Catalog # DD-90-S-BLK0), or equivalent
- Syringe filters, 0.22 µm (Cole-Parmer, Catalog # UX-81053-14), or equivalent
Equipment
- 37 °C, 5% (or 10%) CO2 humidified incubator
- 37 °C water bath
- Autoclave
- Centrifuge
- Dissecting microscope
- Dissection pins, 0.1 mm diameter (Living Systems Instrumentation, Catalog # PIN-0.1MM), or equivalent
- Dissection tools
- Dumont, #5, mirror finish forceps (quantity 2)
- Fine forceps, #5, straight
- Fine scissors, ToughCut®
- Graefe forceps
- Surgical scissors, small
- Vannas-Tübingen spring scissors, 2.5 mm cutting edge
- Hemocytometer
- Inverted microscope
- Laminar flow cell culture hood
- Pipettes
Reagent Preparation
Note: All prepared solutions should be sterile filtered in a laminar flow cell culture hood prior to addition to the medium.
T3 Solution (4 µg/mL)
- Prepare an 8 mg/mL T3 solution in 0.1 N NaOH.
- Add 10 µL of the 8 mg/mL T3 solution to 20 mL of DPBS
- Filter using a 0.22 µm syringe filter to sterilize.
Culture Media
- Add the following reagents to the Neurobasal® medium at the given final concentration.
- In a laminar flow cell culture hood, sterile filter the solution using a 0.22 µm sterile filter unit.
- Add the following growth factors to the solution at the given final concentration.
Procedure
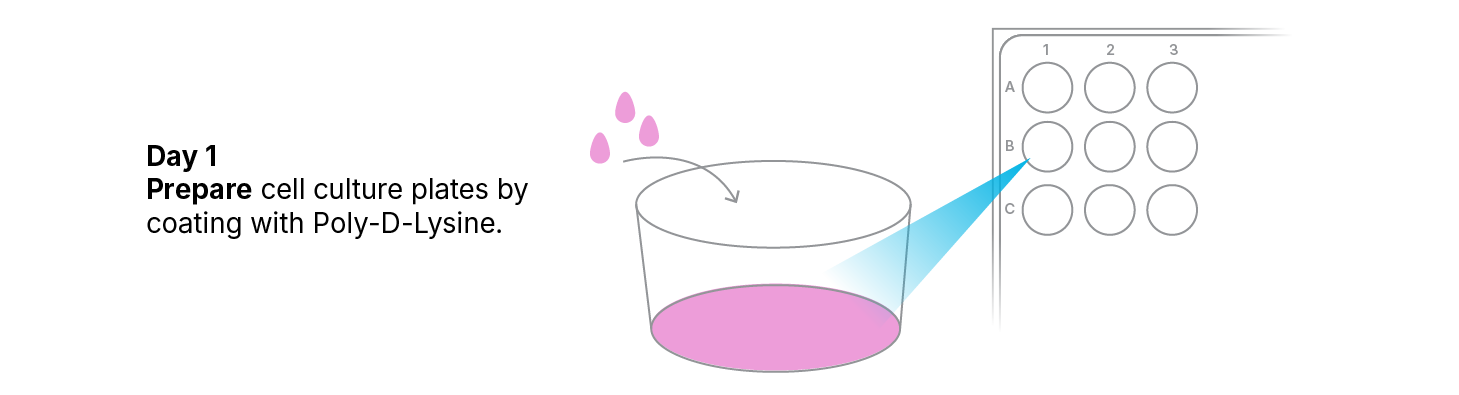
Coating of Cell Culture Plates
Note: Preparation of the cell culture plates should be done in a laminar flow cell culture hood.
- Dilute the Cultrex® Poly-D-Lysine solution with sterile dH2O to a final concentration of 50 µg/mL.
- Add 200 µL of the 50 µg/mL Cultrex® Poly-D-Lysine solution to each well of the cell culture plates. Tilt the plates gently to ensure even coating of the well surface.
- Incubate plates for 1 hour in a 37 °C, 5% CO2 humidified incubator.
- Aspirate the poly-D-lysine solution. Wash the wells three times with sterile dH2O. After the third wash, aspirate the wells to completely remove all liquids.
- Wrap plates with parafilm® to seal. Store plates at 2–8 °C for up to 2 weeks.
Note: Alternatively, spinal motor neurons can be cultured on pre-coated glass coverslips, such as mouse Laminin I and poly-D-lysine coated glass coverslips, 12 mm diameter, # 1.5 thickness (neuVitro, Catalog # GG-12-1.5-laminin), or equivalent. Place one coverslip into each well of a 24-well cell culture plate.
Dissection of Embryonic Rat Spinal Cords
Note: Autoclave dissection tools to sterilize.
- Warm an appropriate amount of media in a 37 °C water bath.
- Place sterile DPBS on ice.
- Asphyxiate pregnant rat with CO2. Recover embryos via cesarean section using the fine scissors and Graefe forceps. Place the embryos in a 100 × 20 mm petri dish containing cold DPBS. Keep the dish on ice.
- Remove the embryos from their individual placenta sacs and wash with cold DPBS.
- Place cleaned embryos in a new 100 × 20 mm petri dish containing cold DPBS. Carefully decapitate each embryo at the head/neck junction using a small surgical scissors. Discard the heads.
- Place the body of the embryo with the ventral side down in a 93 × 22 mm Sylgard®-lined dissection petri dish. Position the embryo so that the tail is pointed towards you. Place a 0.1 mm dissection pin through each limb to fasten the embryo to the dish.
- With the ToughCut® fine scissors, cut off the tail. Under a dissecting microscope, carefully remove skin and tissue with the #5 fine forceps, moving in a ventral direction, until the dorsal surface of the spinal cord is visible.
- Moving caudal to rostral, cut along the dorsal midline of the spinal cord with the Vannas-Tübingen spring scissors to “open” the spinal cord, separating the left and right sides.
- Use the Dumont, #5, mirror finish forceps to remove the extraneous tissue that surrounds the spinal cord, exposing the dorsal root ganglia (DRG). Rub the Dumont, #5, mirror finish forceps between the spinal cord and DRG and meninges to remove these tissues. Repeat this process on the other side to free the spinal cord.
- Remove the spinal cord from the body by grapping onto one end of the spinal cord and lifting it up and away from the body. Use the Vannas-Tübingen spring scissors to cut along the dorsal-ventral midline to trim off the dorsal part of the spinal cord.
- While holding the remaining ventral spinal cord, cut along the ventral midline to bisect it.
- Transfer the isolated ventral spinal cord to a clean 60 × 15 mm dish containing cold L15 medium. Mince the spinal cord into small pieces with the Vannas-Tübingen spring scissors.
Dissociation and Culture of Embryonic Rat Spinal Motor Neurons
Note: From this point forward, any opening of tubes/plates that contain any tissue, cells, media, or reagents should be done in a laminar flow cell culture hood.
- Transfer the dissected spinal cord tissue and L15 medium to a 15 mL conical tube. Centrifuge at 193 × g for 3 minutes at room temperature. Decant the medium from the pelleted tissue pieces.
- Add 3 mL of the TrypZean™ solution, diluted 1:1 in sterile DPBS, to the pelleted tissue. Incubate for 15 minutes in a 37 °C water bath, agitating every 3 minutes.
- Add 3 mL of FBS to the 15 mL conical tube. Centrifuge at 193 × g for 3 minutes at room temperature. Decant the solution from the pelleted tissue pieces.
- Coat a fire-polished Pasteur pipette with FBS.
Add 6 mL of L15 medium to the 15 mL conical tube. Gently triturate the tissue pieces with the fire-polished Pasteur pipette until the solution is homogenous.
Note: Avoid generating bubbles while triturating.
- Prepare a 9% OptiPrep™ solution in L15 medium. Transfer 3 mL of the solution to each of six 15 mL conical tubes.
Divide the homogenized solution evenly among the six tubes containing the OptiPrep™ solution. Centrifuge the tubes at 430 × g for 15 minutes at room temperature.
Note: Brakes should be off during this centrifugation step.
- Carefully collect the top 2 mL of solution from each tube and pool into a 50 mL conical tube. Fill the 50 mL conical tube with L15 medium. Centrifuge at 193 × g for 5 minutes at room temperature. Decant the solution from the pelleted cells.
- Resuspend the cells in 6 mL of L15 medium. Transfer the cell suspension to a new 15 mL conical tube containing 1 mL of 4% BSA. Slowly layer the cell suspension on top of the 4% BSA solution. Centrifuge at 260 × g for 10 minutes at room temperature. Decant the solution from the pelleted cells.
- Resuspend the cells in 250–500 mL of culture media. Mix 10 µL of the cell suspension with 10 µL of 0.4% Trypan blue. Count the live cells.
Cover the previously prepared coverslips with 100 µL of culture media per coverslip. Add 10–20 µL of the cell suspension to each coverslip.
Note: Dissociated cells are plated at a low density (~ 25–50 cells/mm2).
- Incubate the cell culture plates in a 37 °C, 5% CO2 humidified incubator for at least 2 hours to allow the cells to adhere to the coverslips.
- Carefully add 900 µL of culture media to each well of the cell culture plate.
Keep cultured spinal motor neurons in a 37 °C, 5% CO2 humidified incubator until use.
Note: If interested in immunopanning to increase the purity of the motor neuron cultures, refer to Graber, D.J. and B.T. Harris1 for more information.
Exchanging Media in Spinal Motor Neuron Cultures
Note: For long-term cultures, add 100 nM of cytosine arabinoside to the spinal motor neuron cultures on days 4 and 8 to reduce the proliferation of residual astrocytes. Healthy cultures can be maintained for up to 4 weeks.
- Warm an appropriate amount of culture media in a 37 °C, 5% CO2 humidified incubator.
Gently remove half the volume of media (i.e. 50 µL) from each well of the cell culture plates. Gently add 500 µL of new, warmed culture media to each well of the cell culture plates.
Note: Do not remove all the media from the wells of the cell culture plate as this will stress the spinal motor neurons.
- Exchange the culture media every 3-4 days.
References
- Graber, D.J. and B.T. Harris (2013) Cold Spring Harb. Protoc. 2013:319.
- Leach, M.K. et al. (2011) J. Vis. Exp. 48:2389.
- Fantetti, K.N. and D.M. Fekete (2011) J. Vis. Exp. 58:3600.