Human Lysosomal alpha-Glucosidase Antibody (2489B)
R&D Systems | Catalog # MAB83291
Recombinant Monoclonal Antibody.

Key Product Details
Species Reactivity
Human
Applications
Immunohistochemistry, Western Blot
Label
Unconjugated
Antibody Source
Recombinant Monoclonal Rabbit IgG Clone # 2489B
Loading...
Product Specifications
Immunogen
Human embryonic kidney cell HEK293-derived human Lysosomal alpha -Glucosidase
Ala70-Cys952
Accession # P10253
Ala70-Cys952
Accession # P10253
Specificity
Detects human Lysosomal alpha -Glucosidase in direct ELISAs. Detects only 76Kda cleaved band.
Clonality
Monoclonal
Host
Rabbit
Isotype
IgG
Scientific Data Images for Human Lysosomal alpha-Glucosidase Antibody (2489B)
Detection of Human Lysosomal alpha ‑Glucosidase by Western Blot.
Western blot shows lysates of T47D human breast cancer cell line and human peripheral blood mononuclear cells (PBMC). PVDF membrane was probed with 1 µg/mL of Rabbit Anti-Human Lysosomal a-Glucosidase Monoclonal Antibody (Catalog # MAB83291) followed by HRP-conjugated Anti-Rabbit IgG Secondary Antibody (Catalog # HAF008). Specific bands were detected for Lysosomal a-Glucosidase at approximately 75 kDa and 105 kDa (as indicated). This experiment was conducted under reducing conditions and using Immunoblot Buffer Group 1.
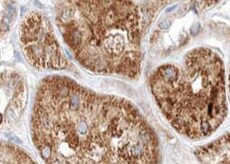
Lysosomal alpha ‑Glucosidase in Human Kidney.
Lysosomal a-Glucosidase was detected in immersion fixed paraffin-embedded sections of human kidney using Rabbit Anti-Human Lysosomal a-Glucosidase Monoclonal Antibody (Catalog # MAB83291) at 3 µg/mL for 1 hour at room temperature followed by incubation with the Anti-Rabbit IgG VisUCyte™ HRP Polymer Antibody (Catalog # VC003). Before incubation with the primary antibody, tissue was subjected to heat-induced epitope retrieval using Antigen Retrieval Reagent-Basic (Catalog # CTS013). Tissue was stained using DAB (brown) and counterstained with hematoxylin (blue). Specific staining was localized to cytoplasm. View our protocol for IHC Staining with VisUCyte HRP Polymer Detection Reagents.Applications for Human Lysosomal alpha-Glucosidase Antibody (2489B)
Application
Recommended Usage
Immunohistochemistry
3-25 µg/mL
Sample: Immersion fixed paraffin-embedded sections of human kidney
Sample: Immersion fixed paraffin-embedded sections of human kidney
Western Blot
1 µg/mL
Sample: T47D human breast cancer cell line and Human peripheral blood mononuclear cells (PBMC)
Sample: T47D human breast cancer cell line and Human peripheral blood mononuclear cells (PBMC)
Reviewed Applications
Read 1 review rated 5 using MAB83291 in the following applications:
Formulation, Preparation, and Storage
Purification
Protein A or G purified from cell culture supernatant
Reconstitution
Reconstitute at 0.5 mg/mL in sterile PBS. For liquid material, refer to CoA for concentration.
Loading...
Formulation
Lyophilized from a 0.2 μm filtered solution in PBS with Trehalose. *Small pack size (SP) is supplied either lyophilized or as a 0.2 µm filtered solution in PBS.
Shipping
Lyophilized product is shipped at ambient temperature. Liquid small pack size (-SP) is shipped with polar packs. Upon receipt, store immediately at the temperature recommended below.
Stability & Storage
Use a manual defrost freezer and avoid repeated freeze-thaw cycles.
- 12 months from date of receipt, -20 to -70 °C as supplied.
- 1 month, 2 to 8 °C under sterile conditions after reconstitution.
- 6 months, -20 to -70 °C under sterile conditions after reconstitution.
Calculators
Background: Lysosomal alpha-Glucosidase
References
- Hoefsloot L.H. et al. (1988) EMBO J. 7:1697.
- Wan, L. et al. (2008) J. Neurol. 255:831.
- Fukuda, T. et al. (2007) Curr. Neurol. Neurosci. Rep. 7:71.
- Van Gelder, C.M. et al. (2014) J Inherit Metab Dis. In press.
- Toscano, A. and Schoser, B. (2013) J. Neurol. 260:951.
Long Name
Glucosidase, Alpha; Acid
Alternate Names
Acid alpha-Glucosidase, Acid Maltase, GAA, LYAG, Lysosomal alphaGlucosidase
Gene Symbol
GAA
UniProt
Additional Lysosomal alpha-Glucosidase Products
Product Documents for Human Lysosomal alpha-Glucosidase Antibody (2489B)
Certificate of Analysis
To download a Certificate of Analysis, please enter a lot or batch number in the search box below.
Note: Certificate of Analysis not available for kit components.
Product Specific Notices for Human Lysosomal alpha-Glucosidase Antibody (2489B)
For research use only
Related Research Areas
Customer Reviews for Human Lysosomal alpha-Glucosidase Antibody (2489B) (1)
5 out of 5
1 Customer Rating
Have you used Human Lysosomal alpha-Glucosidase Antibody (2489B)?
Submit a review and receive an Amazon gift card!
$25/€18/£15/$25CAN/¥2500 Yen for a review with an image
$10/€7/£6/$10CAN/¥1110 Yen for a review without an image
Submit a review
Customer Images
Showing
1
-
1 of
1 review
Showing All
Filter By:
-
Application: ImmunohistochemistrySample Tested: Kidney tissueSpecies: HumanVerified Customer | Posted 06/27/2022

There are no reviews that match your criteria.
Protocols
Find general support by application which include: protocols, troubleshooting, illustrated assays, videos and webinars.
- Antigen Retrieval Protocol (PIER)
- Antigen Retrieval for Frozen Sections Protocol
- Appropriate Fixation of IHC/ICC Samples
- Cellular Response to Hypoxia Protocols
- Chromogenic IHC Staining of Formalin-Fixed Paraffin-Embedded (FFPE) Tissue Protocol
- Chromogenic Immunohistochemistry Staining of Frozen Tissue
- ClariTSA™ Fluorophore Kits
- Detection & Visualization of Antibody Binding
- Fluorescent IHC Staining of Frozen Tissue Protocol
- Graphic Protocol for Heat-induced Epitope Retrieval
- Graphic Protocol for the Preparation and Fluorescent IHC Staining of Frozen Tissue Sections
- Graphic Protocol for the Preparation and Fluorescent IHC Staining of Paraffin-embedded Tissue Sections
- Graphic Protocol for the Preparation of Gelatin-coated Slides for Histological Tissue Sections
- IHC Sample Preparation (Frozen sections vs Paraffin)
- Immunofluorescent IHC Staining of Formalin-Fixed Paraffin-Embedded (FFPE) Tissue Protocol
- Immunohistochemistry (IHC) and Immunocytochemistry (ICC) Protocols
- Immunohistochemistry Frozen Troubleshooting
- Immunohistochemistry Paraffin Troubleshooting
- Preparing Samples for IHC/ICC Experiments
- Preventing Non-Specific Staining (Non-Specific Binding)
- Primary Antibody Selection & Optimization
- Protocol for Heat-Induced Epitope Retrieval (HIER)
- Protocol for Making a 4% Formaldehyde Solution in PBS
- Protocol for VisUCyte™ HRP Polymer Detection Reagent
- Protocol for the Preparation & Fixation of Cells on Coverslips
- Protocol for the Preparation and Chromogenic IHC Staining of Frozen Tissue Sections
- Protocol for the Preparation and Chromogenic IHC Staining of Frozen Tissue Sections - Graphic
- Protocol for the Preparation and Chromogenic IHC Staining of Paraffin-embedded Tissue Sections
- Protocol for the Preparation and Chromogenic IHC Staining of Paraffin-embedded Tissue Sections - Graphic
- Protocol for the Preparation and Fluorescent IHC Staining of Frozen Tissue Sections
- Protocol for the Preparation and Fluorescent IHC Staining of Paraffin-embedded Tissue Sections
- Protocol for the Preparation of Gelatin-coated Slides for Histological Tissue Sections
- R&D Systems Quality Control Western Blot Protocol
- TUNEL and Active Caspase-3 Detection by IHC/ICC Protocol
- The Importance of IHC/ICC Controls
- Troubleshooting Guide: Immunohistochemistry
- Troubleshooting Guide: Western Blot Figures
- Western Blot Conditions
- Western Blot Protocol
- Western Blot Protocol for Cell Lysates
- Western Blot Troubleshooting
- Western Blot Troubleshooting Guide
- View all Protocols, Troubleshooting, Illustrated assays and Webinars
Loading...
Associated Pathways
