Neurodegenerative Disease
Neurodegenerative diseases refer to a group of age-associated conditions with progressive loss of neuronal structure and function, often accompanied by aberrant protein accumulation, resulting in cognitive disability, motor deficits, and dementia. Ranging from neurodegenerative disorders such as Alzheimer’s disease to psychiatric conditions such as depression and anxiety-related disorders, diseases that affect nervous system function are some of the most prevalent and least understood diseases.
The four most common neurodegenerative diseases include:
These diseases each have different clinical presentations and affect different neuronal populations and brain areas; however, commonalities identified between the diseases suggest that abnormal protein aggregation, failure of autophagy, mitochondrial dysfunction, and oxidative stress, as well an inflammatory environment and reactive microglia, contribute to the development of all these diseases. Further advances in our understanding of the molecular mechanisms underlying nervous systems dysfunction in these debilitating diseases is critical for the development of effective treatments.
R&D Systems offers a range of research tools to study the biological processes that accompany neurological/neurodegenerative disease.
Most Common Neurodegenerative Diseases and Their Pathologies
| Neurodegenerative Disease | Brain Regions Affected | Pathology | Major Cell Types Affected |
|---|---|---|---|
| Alzheimer's disease |
|
|
|
| Parkinson's disease |
|
|
|
| Huntington's disease |
|
|
|
| Amyotrophic lateral sclerosis |
|
|
|
Alzheimer’s Disease
Alzheimer’s disease (AD) is marked by a progressive cognitive impairment including memory loss and behavioral changes. Numerous environmental and generic risk factors have been associated with developing this generally sporadic disease. AD is characterized by amyloid-beta accumulation in extracellular neural tissues, as well as intracellular neurofibrillary tangles consisting of phosphorylated tau protein. The result is a deterioration of cholinergic neurons and loss of synaptic function in the hippocampus and cerebral cortex.
Major Protein Targets Involved in AD
| Gene | Protein Target | Role in Disease |
|---|---|---|
| APP | Amyloid precursor protein | Integral membrane protein that is sequentially processed by beta-secretase and gamma-secretase to produce amyloid-beta that can form oligomers and lead to amyloid plaque formation. |
| PSEN1/PSEN2 | Facilitate gamma-secretase cleavage of APP. Mutations in presenilins increase production of the aggregation prone amyloid-beta variant (Aβ42). | |
| APOE/APOE4 | Apolipoprotein E | Major cholesterol carrier, supports lipid transport. ApoE isoforms differentially regulate amyloid-beta aggregation and clearance in the brain. The ApoE ε4 allele is associated with increased risk of developing Alzheimer's disease. |
| MAPT | Tau | A neuronal microtubule associated protein found in axons. Promotes tubulin polymerization and stabilizes microtubules. Hyperphosphorylated tau is a major component of neurofibrillary tangles. |
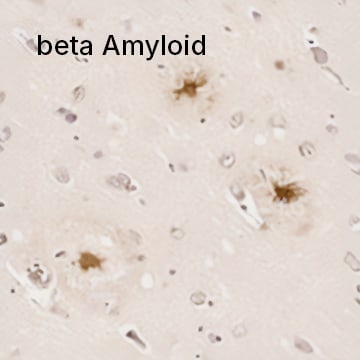
beta Amyloid Antibody (MOAB-2) [NBP2-13075] - IHC analysis of a formalin fixed paraffin embedded tissue section of human brain (Alzheimer's disease, hippocampus) using 1:200 dilution of anti-beta Amyloid antibody (clone MOAB-2). The staining was developed with HRP labeled anti-mouse secondary antibody and DAB reagent, and nuclei of cells were counter-stained with Hematoxylin. This beta Amyloid antibody specifically stained the cells with Abeta 42/ Abeta aggregates.
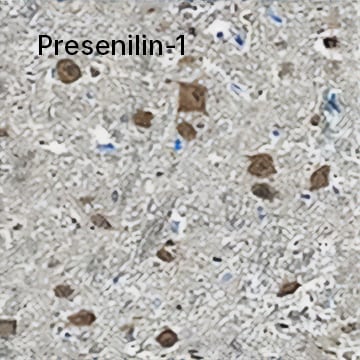
Presenilin-1 was detected in immersion fixed paraffin-embedded sections of human brain (medulla) using Mouse Anti-Human Presenilin-1 N-Terminal Fragment Monoclonal Antibody (Catalog # MAB149) at 1.7 µg/mL for 1 hour at room temperature followed by incubation with the Anti-Mouse IgG VisUCyte™ HRP Polymer Antibody. Tissue was stained using DAB (brown) and counterstained with Hematoxylin. Specific staining was localized to neuronal cell bodies.
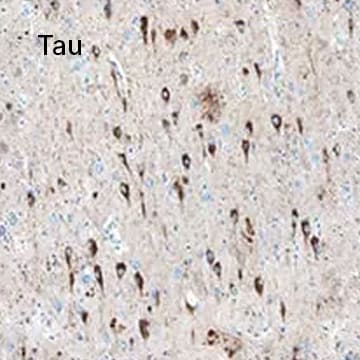
Tau was detected in immersion fixed paraffin-embedded sections of human Alzheimer's brain using Mouse Anti-Tau Monoclonal Antibody (Catalog # MAB3494) at 15 µg/mL overnight at 4 °C. Before incubation with the primary antibody, tissue was subjected to heat-induced epitope retrieval using Antigen Retrieval Reagent-Basic. Tissue was stained using the Anti-Mouse HRP-DAB Cell & Tissue Staining Kit (brown) and counterstained with Hematoxylin. Specific staining was localized to neuronal cell bodies and processes.
Parkinson’s Disease
Parkinson’s disease (PD) is a common neurodegenerative disease characterized by the loss of midbrain dopaminergic neurons, mainly in the substantia nigra. It is defined by the presence of alpha-synuclein aggregates (Lewy bodies) and accumulation of defective mitochondria, leading to neuronal death. PD patients display a variety of motor symptoms such as resting tremors, rigidity and postural instability, along with non-motor symptoms including depression, sleep disorder and olfactory dysfunction. Sporadic forms of the disease constitute more than 90% of cases and the onset occurs over the age of 60. Currently, there is no cure for PD, but its motor symptoms can be alleviated temporarily with dopamine inducing approaches. New therapies concentrate on neuroprotective and disease-modifying strategies.
Major Protein Targets Involved in PD
| Gene | Protein Target | Role in Disease |
|---|---|---|
| LRRK2 | Leucine rich repeat kinase 2 | Most commonly known genetic contributor to PD. Mutations are linked to aberrant inflammatory responses, increased oxidative stress, synaptic dysfunction, abnormal autophagy functions, and more. |
| PINK | PTEN-induced putative kinase 1 | A mitochondrial serine/threonine protein kinase with a protective function against stress-induced mitochondrial dysfunction. Targets defective mitochondria for degradation. Mutations in PINK1 lead to accumulation of misfolded proteins in mitochondria. |
| TH | Tyrosine hydroxylase | Enzyme that catalyzes the hydroxylation of L-tyrosine to L-DOPA, a critical dopamine precursor. |
| PARK2 | Parkin | Plays a role in the ubiquitin-mediated proteolytic pathway. Loss of parkin E4 ubiquitin ligase function contributes to accumulation of toxic and/or misfolded proteins. |
| PARK7 | Protein deglycase DJ-1 | Various roles in transcriptional regulation, oxidative stress response, and mitochondrial function. Senses cellular redox metabolism and scavenges free radicals. Mutations associated with early-onset familial forms of PD. |
| SNCA | alpha-Synuclein | Involved in the regulation of synaptic transmission and neurotransmitter release. Aggregates of α-synuclein correlate with degeneration of dopaminergic neurons. Main component of Lewy bodies. |
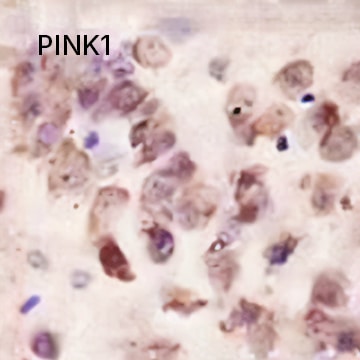
PINK1 Antibody [NBP1-49678] - IHC stain in paraffin-embedded mouse brain.
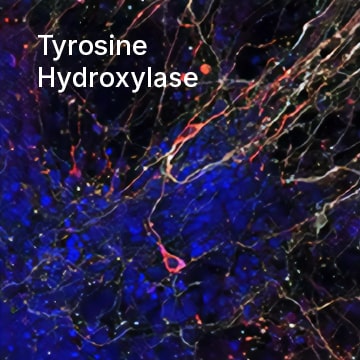
Tyrosine Hydroxylase was detected in immersion fixed mouse embryonic stem cells differentiated into dopaminergic neurons using Mouse Anti-Human/Mouse Tyrosine Hydroxylase Monoclonal Antibody (Catalog # MAB7566) at 10 µg/mL for 3 hours at room temperature. Cells were stained using the NorthernLights™ 557-conjugated Anti-Mouse IgG Secondary Antibody (red; Catalog # NL007). Cells were double stained using the NorthernLights™ 637-conjugated Mouse Anti-Neuron-specific beta-III Tubulin Monoclonal Antibody (white; Catalog # NL1195V). Cells were counterstained with DAPI (blue). Specific staining of Tyrosine Hydroxylase was localized to cytoplasm of dopaminergic neurons.
Parkinson's Disease-Related Targets
| 14-3-3 Proteins | ||
| 5-HT Receptors | ||
| Acetylcholine Receptors | ||
| Adenosine Receptors | ||
| Adrenergic Receptors | ||
| Autophagy | ||
| Bcl-2 Family | ||
| Calcium Channels | ||
| Caspases | ||
| Catechol O-Methyltransferase (COMT) | ||
| Cyclooxygenases (COX) | ||
| Deubiquitinating Enzymes | ||
| Dopamine Receptors | ||
| Dopamine Transporters | ||
| Dynamin | ||
| Ferroptosis | ||
| GABAA and GABAA-ρ Receptors | ||
| GABAB Receptors | ||
| Glutamate Dehydrogenases | ||
| Glutamate Receptors | ||
| Glycosylases | ||
| Heat Shock Proteins | ||
| LRRK2 | ||
| Mitofusins | ||
| Monoamine Oxidases | ||
| Neural Stem Cells | ||
| Neurofilaments | ||
| Neuronal Metabolism | ||
| Oxidative Phosphorylation | ||
| Phosphodiesterases | ||
| Poly(ADP-ribose) Polymerase | ||
| Protein Arginine Methyltransferases | ||
| Sigma Receptors | ||
| Sirtuins | ||
| Stearoyl-CoA 9-Desaturase (SCD-1) | ||
| Trace Amine 1 Receptor | ||
| Trk Receptors | ||
| Vesicular Monoamine Transporters (VMAT) | ||
| Other Parkinson's Disease Related Targets | ||
Huntington’s Disease
Huntington’s disease (HD) is marked by a progressive degeneration of neurons of the striatum and cortex. The disease is familial and follows an autosomal dominant inheritance pattern. It is characterized by a polyglutamine repeat expansion in the huntingtin protein that results in formation of neuronal inclusions and subsequent neuronal loss. This leads to movement, psychiatric and cognitive impairments. Patients are usually diagnosed at 40 years of age and have life expectancy of around 15 years. Currently, no cure exists for the disease.
Major Protein Targets Involved in HD
| Gene | Protein Target | Role in Disease |
|---|---|---|
| HTT | Huntingtin | Mutant huntingtin protein forms aggregates and has deleterious effects on neuronal gene transcription, axonal transport and mitochondrial function. |
| HAP1 | Huntingtin associated protein 1 | A cytoplasmic protein, interacts with Huntingtin, among other targets in regulating microtubule-dependent transport, vesicular trafficking, nerve growth and transcriptional regulation. |
| SOD1 | Superoxide dismutase | An antioxidant enzyme involved in the defense against reactive oxygen species. |
| BDNF | Brain-derived neurotrophic factor | Pro-survival factor in neurons, required for axonal transport and neuronal survival. Mutant HTT decreases the levels of BDNF leading to reduced neuronal survival. |
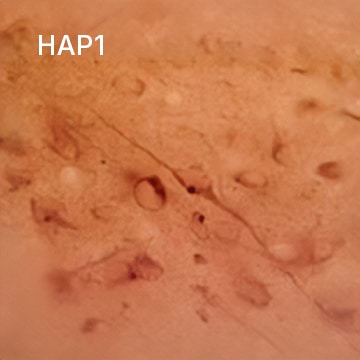
HAP1 Antibody (1B6) [NB110-74569] - Staining of rat hypothalamus.
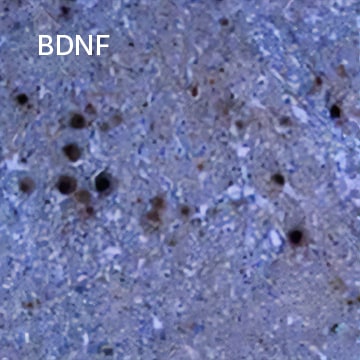
BDNF was detected in immersion fixed paraffin-embedded sections of human spinal cord using Chicken Anti-Human BDNF Antigen Affinity-purified Polyclonal Antibody (Catalog # AF248) at 15 µg/mL overnight at 4 °C. Tissue was stained using HRP-DAB detection (brown) and counterstained with hematoxylin (blue).
Amyotrophic Lateral Sclerosis
Amyotrophic Lateral Sclerosis (ALS; commonly known as Lou Gehrig's disease) is a disease of degeneration of motor neurons in the brain and the spinal cord. It is the third most common neurodegenerative disease and the most common motor neuron disease. Symptoms include loss of control of muscle movement as well as muscle weakness, atrophy, twitching and cramps. The onset of the disease occurs at approximately 60 years old and patients survive on average three years from diagnosis. Most cases of ALS are sporadic and involve a number of genes, with further genes still likely to be discovered. However, 60% of familial ALS can be attributed to mutations in four genes: SOD1, TDP-43/TARDBP, FUS and C9orf72. Aberrant autophagy and RNA metabolism are two processes heavily implicated in disease progression. Protein aggregates are also a common feature of ALS.
Major Protein Targets Involved in ALS
| Gene | Protein Target | Role in Disease |
|---|---|---|
| SOD1 | Superoxide dismutase | An antioxidant enzyme involved in the defense against reactive oxygen species. Mutations in SOD1 are linked to familial ALS. |
| TARDBP | TDP-43/TARDBP | Nuclear RNA/DNA binding phosphoprotein that regulates transcription and splicing in the cell nucleus. Mutations in TDP-43 cause its aggregation within inclusion bodies in the cytoplasm of motor neurons. |
| FUS | FUS | Mutant variants are involved in abnormal mRNA processing and form aggregates in motor neurons. |
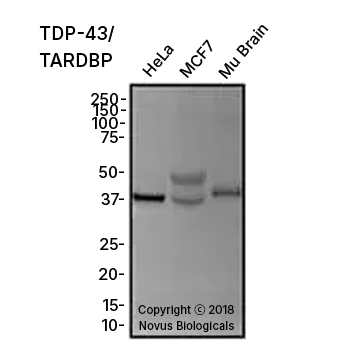
TDP-43/TARDBP Antibody [NB110-55376] - Total protein from HeLa, MCF7 and mouse brain was separated on a 12% gel by SDS-PAGE, transferred to PVDF membrane and blocked in 5% non-fat milk in TBST. The membrane was probed with 1.0 μg/mL anti-TARDBP in 1% block buffer and detected with an anti-rabbit HRP secondary antibody using chemiluminescence.
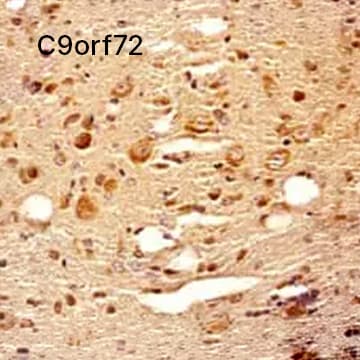
C9orf72 Antibody [NBP2-47146] - IHC analysis of a formalin fixed paraffin embedded tissue section of mouse brain using C9orf72 antibody (NBP2-47146) at 1:300 dilution with HRP-DAB detection and Hematoxylin (blue) counterstaining. The antibody generated strong/specific cytoplasmic signal in the neurons and other cell types in the tested section.
ALS-Related Targets
| Dynactins | ||
| Glutamate Transporters (EAAT) | ||
| Sirtuins | ||
| Superoxide Dismutase (SOD) | ||
| Synucleins | ||
| Other ALS-Related Targets | ||
Multiple Sclerosis
Myelin is the insulating sheath that surrounds axons and facilitates rapid transmission of electrical signals between neurons. Myelin is composed of lipid, myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), and proteolipid protein (PLP). Degeneration of the myelin sheath occurs during some auto-immune diseases, such as multiple sclerosis and acute disseminated encephalomyelitis.
Multiple Sclerosis-Related Targets
| Acid-Sensing Ion Channels | ||
| ASK1 | ||
| Calcium-Activated Potassium (KCa) Channels | ||
| Cannabinoid Receptors | ||
| Chemokine Receptors | ||
| Complement | ||
| Cytokines | ||
| Glutamate (Ionotropic) Receptors | ||
| GPCRs | ||
| Immunosuppressants | ||
| Integrins | ||
| Interleukins | ||
| Inward Rectifier Potassium (Kir) Channels | ||
| ITK | ||
| Na+/Ca2+ Exchanger | ||
| Nogo | ||
| Nrf2 | ||
| Retinoic Acid (RAR) and Related Receptors | ||
| Retinoid X Receptors | ||
| ROS and Other Gaseous Donors | ||
| Sphingokinases | ||
| STATs | ||
| Tankyrase | ||
| TRPM Channels | ||
| TSPANs | ||
| Vitamin D Receptors | ||
| Voltage-gated Potassium (KV) Channels | ||
| Wnt Signaling | ||
| Additional Multiple Sclerosis Related Targets | ||
Literature for Neurodegenerative Disease Research
Alzheimer's Disease Research Product Guide

Learn more about Alzheimer's disease and how R&D Systems' products can advance your research in our Alzheimer's Disease Research Product Guide.
Parkinson's Disease Research Product Guide

Explore the mechanisms behind Parkinson's disease (PD), and browse the relevant, high-quality reagents available from R&D Systems.
Huntington's Disease Research Product Guide

Understand the pathology and key molecular targets involved in Huntington's disease (HD) with our Huntington's Disease Research Product Guide.
Alzheimer's Disease Life Science Poster

This poster summarizes the cellular and molecular mechanisms of Alzheimer's disease (AD), a debilitating and progressive neurodegenerative disease.
Parkinson's Disease Life Science Poster

This poster outlines the neurobiology of Parkinson's disease (PD), as well as highlighting current and emerging therapeutic treatments for PD.
Huntington's Disease Life Science Poster

This poster summarizes the pathophysiology of Huntington's disease (HD), as well as highlighting the use of iPSCs for HD modeling.
Cell Structure and Signaling Molecules
Resources for Neurodegenerative Disease Research
- Exosomes in Neurodegenerative Disease
- Fetal Alcohol Syndrome and Apoptosis
- Cytokines - Pieces of the Autism Puzzle
- Chemokine Receptors and Multiple Sclerosis Pathogenesis
- Gene Expression in the Aging Brain
- Mini-review: Multiple Sclerosis: Immune Cell Access to the Central Nervous System
- Oncomodulin: The inflammatory response and nerve regeneration
- Th17 Differentiation: An Evolving Target for Multiple Sclerosis Therapy
- Vitamin D Deficiency as a Risk Factor for Multiple Sclerosis
- Neuroprotective & Pathogenic Effects of VEGF Signaling
- ApoE Suppresses Cyclophilin A to Maintain Cerebrovascular Integrity: A Pharmacological Target for Alzheimer’s Disease
- Astrocytes Secrete Neuroprotective Factors & Protect Against Glutamate-induced Neural Cell Apoptosis
- Neural Activation Triggers CCL20 Expression & Promotes T Cell Entry into the Spinal Cord in a Mouse Model of Multiple Sclerosis
- Alpha-Synuclein-Based Model for Studying Parkinson's Disease Pathology
- TIQ and Parkinson's Disease
- VEGF & Amyotrophic Lateral Sclerosis
- Apoptosis Research
- Adult Neurogenesis Research
- Autoimmunity Poster